Spectrométrie de masse
Localisé au 2ème étage du Centre de Biologie et de Recherche en Santé, le plateau de spectrométrie de masse est ouvert à l’ensemble des laboratoires de l’Université de Limoges ainsi qu’aux structures extérieures, qu’elles soient publiques ou privées.
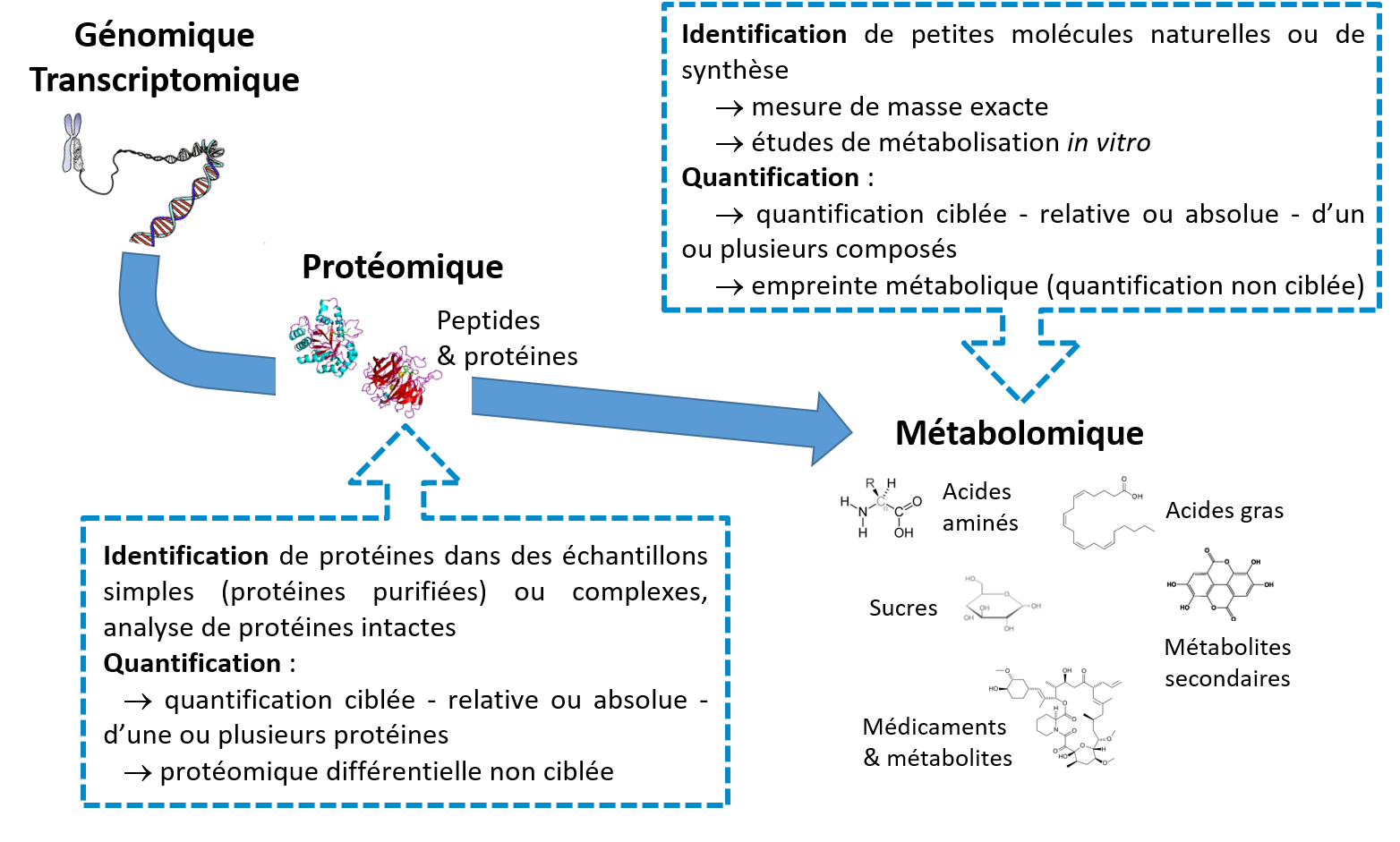
Il dispose d’outils permettant de caractériser (identification et quantification – relative ou absolue) des petites molécules (métabolites, médicaments, …) et des protéines dans des échantillons biologiques ou issus de synthèse chimique.
Personnel du plateau
Emilie PINAULT
|
Spectromètre de masse LCMS8060 (Shimadzu)
|
|
Spectromètre de masse MALDI TOF Axima Confidence (Shimadzu)
|
|
Spectromètre de masse TimsTOF Pro2 (Bruker) couplé
|
||



Les équipements du plateau (TimsTOF Pro2 et MALDI TOF Axima Confidence) permettent la caractérisation de petites molécules dans des échantillons simples (composé purifié ou issu d’une synthèse chimique) ou complexes (extrait).Le plateau propose également une prestation d’étude de métabolisation in vitro de médicaments (incubation avec des microsomes hépatiques ou rénaux) suivie d’une identification par spectrométrie de masse des métabolites ainsi produits.En infusion, après séparation par LC ou après dépôt sur plaque MALDI, la mesure de la masse exacte des composés grâce à la haute résolution (HR-MS) conduit à la suggestion de formules brutes avec une précision inférieure à 10ppm et les profils de fragmentation (MS/MS), véritable « empreinte digitale », sont comparés aux bases de données publiques et aux prédictions de fragmentation obtenues in silico.
|
|

Il est possible de réaliser des analyses sur protéines intactes, pour obtenir des informations de pureté et de poids moléculaire.Cette technique a été appliquée avec succès sur des protéines de 8 kDa (protéines recombinantes) jusqu’à 70 kDa (albumine humaine), ce qui a permis de mettre en évidence des isoformes de ces protéines dues à des modifications post-traductionnelles. |
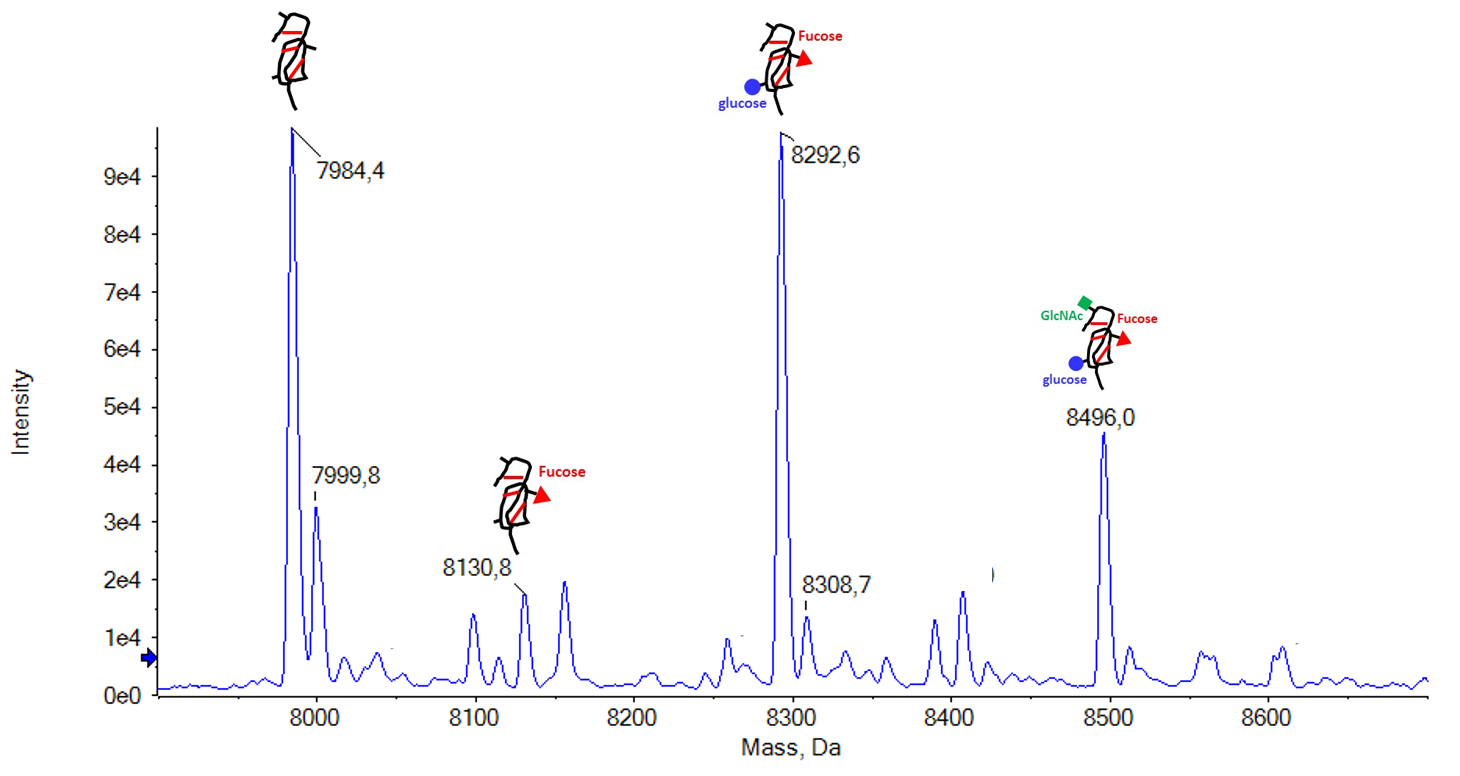
(Pennarubia et al., 2020, Glycobiology) |
Les protéines peuvent être identifiées par séquençage peptidique à partir d’extraits protéiques ou de fluides biologiques, avec ou sans fractionnement ou enrichissement préalable.
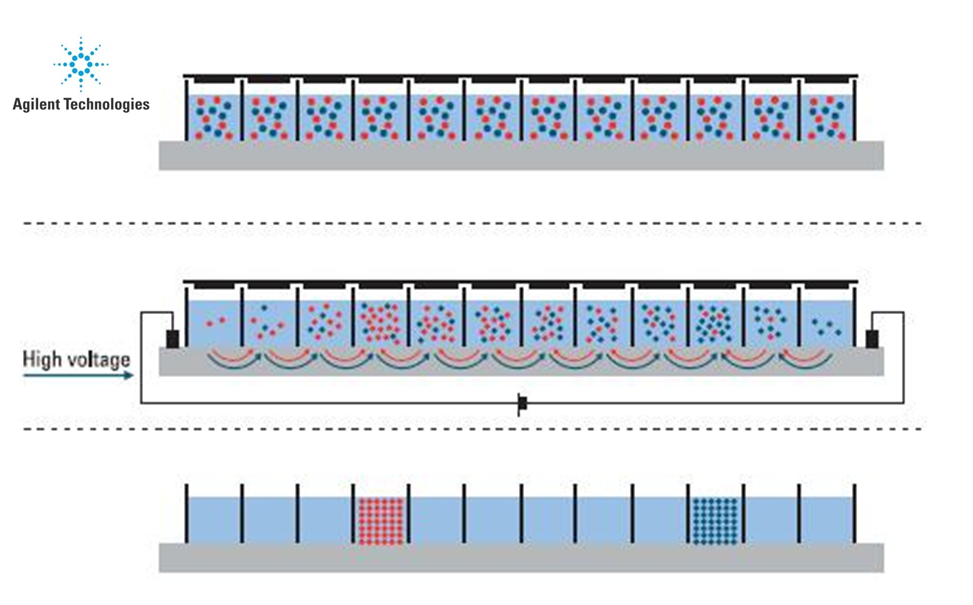
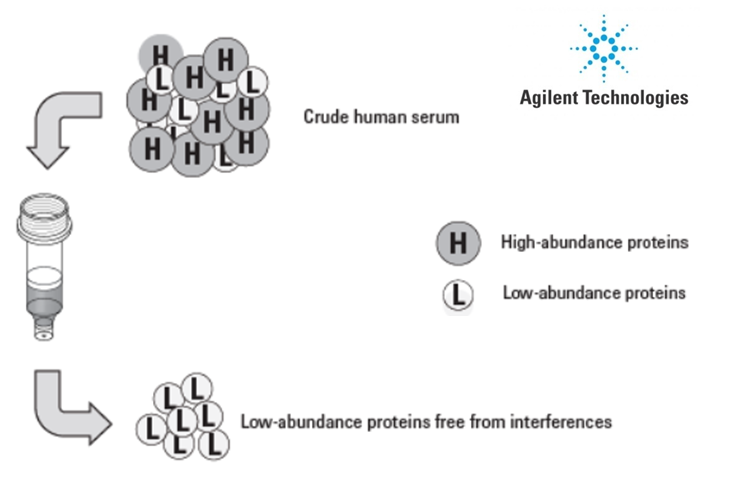
Le plateau dispose de plusieurs techniques de fractionnement (gels SDS-PAGE mono et bidimensionnels ; IsoElectrofocalisation Off Gel) et d’enrichissement (déplétion des 6 protéines majoritaires du plasma humain ; enrichissement ProteoMiner).
|
Gels
|
Isoelectrofocalisation
|
Déplétion des
|
Enrichissement |

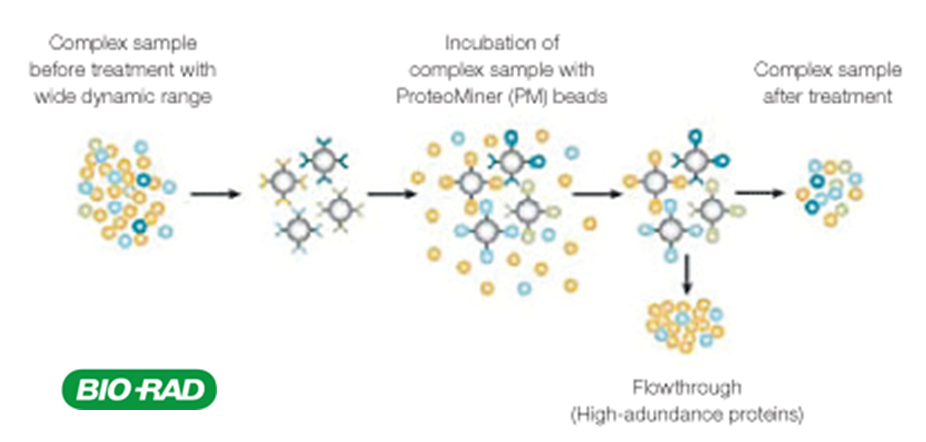
|
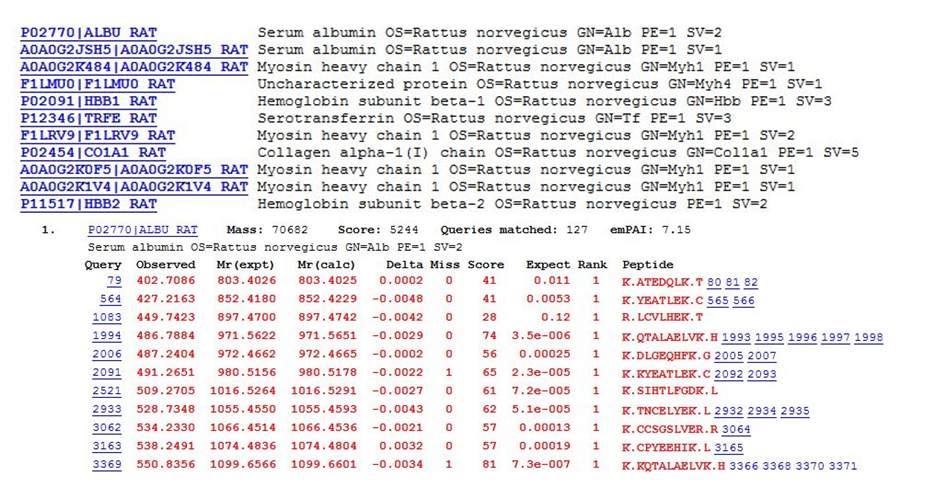
Après une étape obligatoire de digestion des protéines par la trypsine, les peptides sont analysés en mode DDA (Data Dependent Acquisition) par spectrométrie de masse nano-LC QTOF. Les données sont retraitées par les logiciels ProteoScape et/ou Mascot : chaque spectre de fragmentation est interprété pour déterminer la séquence du peptide en acides aminés. L’ensemble des séquences peptidiques est comparé avec les bases de données protéiques (Uniprot, NCBI, …) générant ainsi une liste de protéines identifiées avec leurs peptides. |
 |
||
|
Mascot
|
ProteoScape
|
||

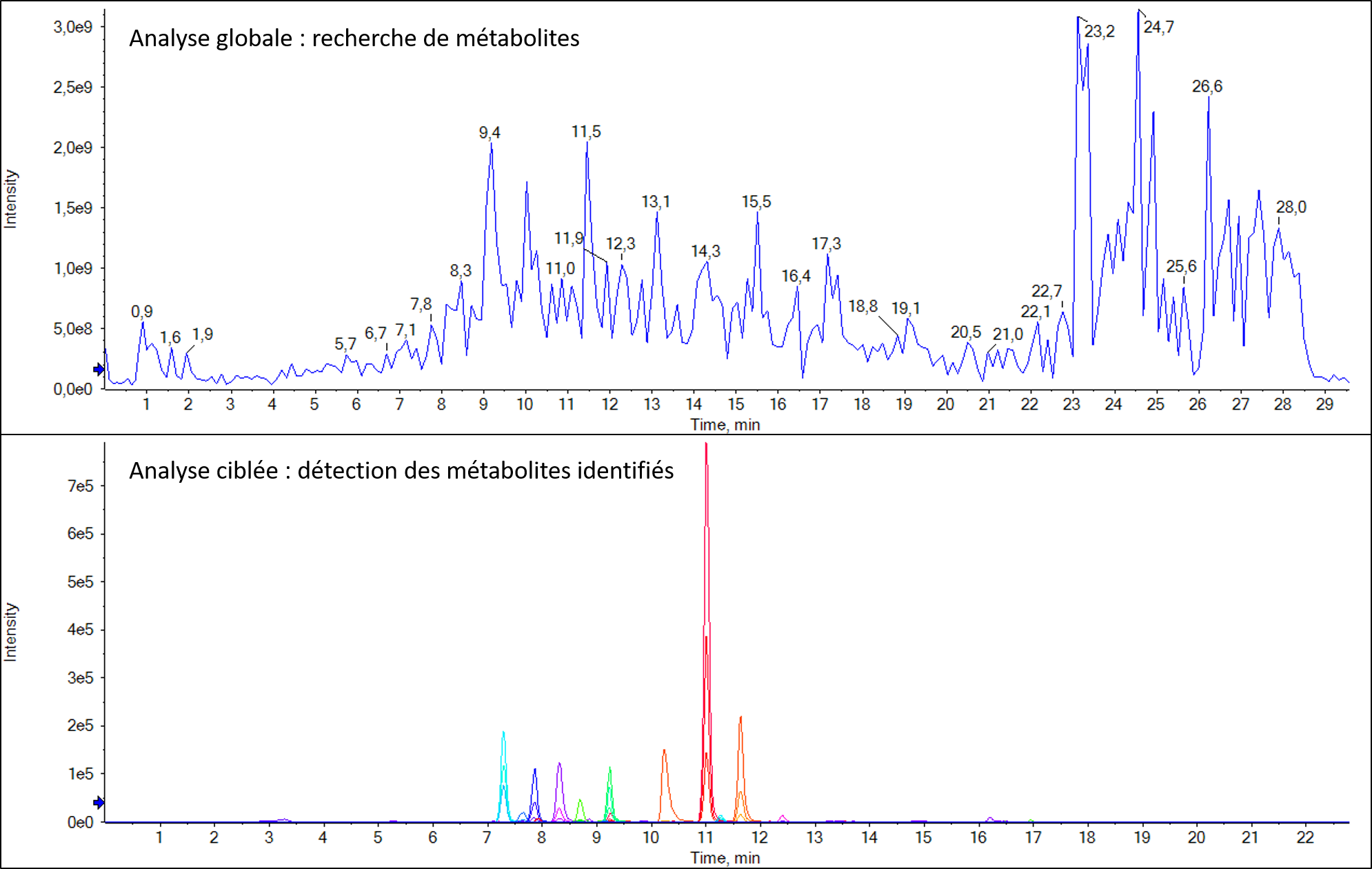
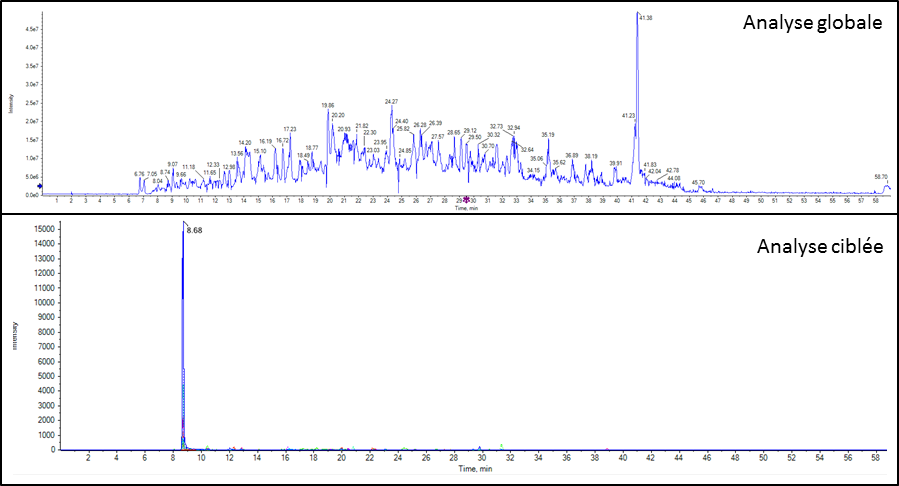
L’analyse en mode MRM (pour Multiple Reaction Monitoring) permet de quantifier de façon relative ou absolue (uniquement si le composé pur est disponble), de un à plusieurs centaines de composés dans un échantillon complexe, qu’il s’agisse de petites molécules ou de protéines.
Dans le cas d’une quantification absolue, il est possible de mettre en place une validation de la méthode analytique (préparation d’échantillon et analyse par spectrométrie de masse) pour prouver que celle-ci est exacte et fidèle. Pour cela, plusieurs paramètres sont évalués : limites de détection et de quantification, linéarité de la gamme, reproductibilité, répétabilité, rendement, dilutions, effet matrice…
Ce type d’analyse requiert de connaître la fragmentation des composés à quantifier (par l’étude du composé pur ou lors d’acquisition LC-MS/MS en mode DDA – voir onglets Identification des petites molécules ou des protéines) pour sélectionner 2 ou 3 fragments représentatifs les plus intenses et spécifiques pour chaque composé qui sont intégrés dans la méthode MRM.
Chaque pic, correspondant à la détection spécifique d’un composé ciblé, est intégré ; l’aire mesurée étant quantitative et comparable de façon relative ou absolue aux dfférentes échantillons.
L’ajout d’un standard interne est vivement conseillé pour normaliser les analyses et éliminer une partie des biais de manipulations et d’analyses. Une version alourdie (D, 13C, 15N) du composé à doser ou un composé de la même famille ou avec des propriétés physico-chimiques proches sont privilégiés.
Exemples :
|
Détection d’un candidat médicament et de ses métabolites dans un échantillon urinaire après extraction QuECHERS
|
Détection d’un peptide dans du plasma après enrichissement ProteoMiner et digestion trypsique
|
La protéomique différentielle DIA (pour Data-Independant Acquisition) permet d’obtenir des informations quantitatives relatives pour toutes les protéines présentes dans une librairie protéique.
Elle présente l’avantage de pouvoir ré-analyser les données DIA acquises autant de fois que nécessaires avec des librairies différentes (par exemple, une librairie contenant uniquement des protéines phosphorylées).
|
La librairie est constituée de l’ensemble des protéines représentatives des échantillons. Pour chaque protéine, sont listés les peptides spécifiques (masse m/z et temps de rétention) et pour chaque peptide, les fragments (m/z et intensité relative). L’acquisition des données est effectuée en mode DIA (Data-Independent Acquisition) : à chaque cycle, le spectre MS (entre m/z 400 et m/z 1250) est découpé en 20 à 40 fenêtres de masse. L’ensemble des pics présents dans chaque fenêtre seront fragmentés, le spectre MS/MS acquis correspond ainsi à la somme des fragments de tous les peptides présents dans cette gamme de masse. |
|

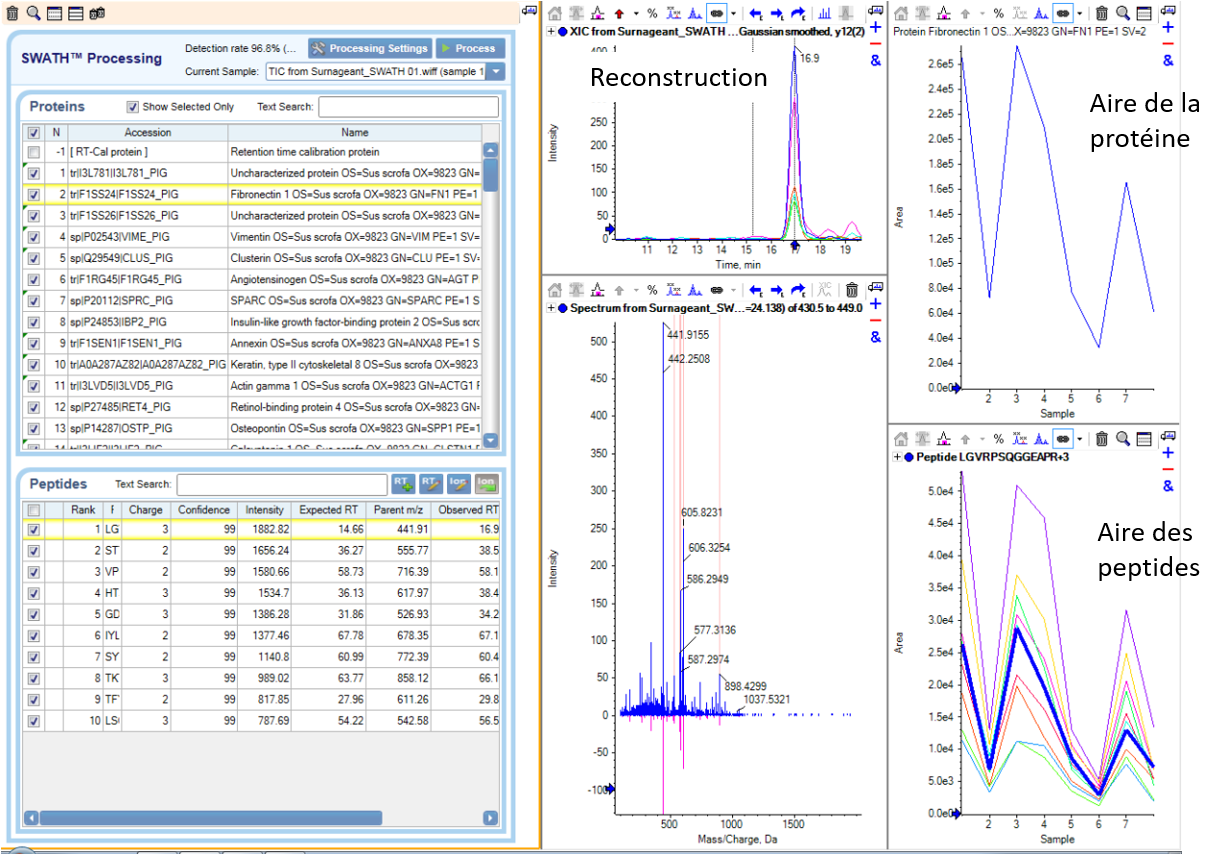
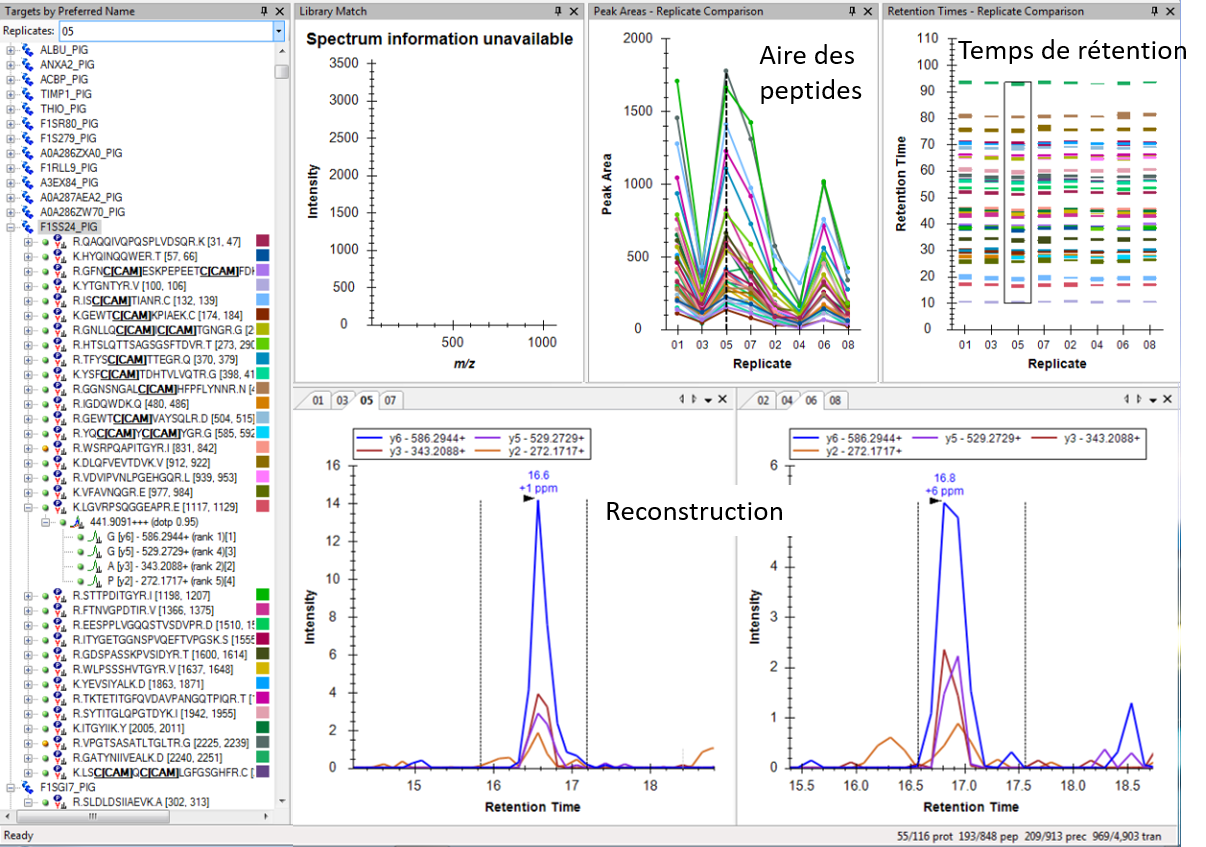
Puis, en comparant les données DIA avec les données de la librairie, le logiciel de retraitement (ProteoScape ou Skyline) permet de ré-attribuer les fragments de chaque peptide. Chaque peptide est quantifié par la mesure de l’aire sous la courbe après reconstruction de l’abondance des différents fragments au cours du temps.
La quantification relative des protéines dans les différentes échantillons sera réalisée par comparaison des abondances correspondant à la somme des aires des peptides spécifiques la constituant.
|
Retraitement via le logiciel PeakView/SWATH2.0
|
Retraitement via le logiciel Skyline
|
|
Le plateau dispose de plusieurs logiciels propriétaires (ProteinPilot, PeakView, MultiQuant, Insight) et multi-plateformes (Sirius, Skyline) pour le retraitement des données issues des spectromètres de masse du plateau. Le retraitement des données peut être pris en charge par l’ingénieur du plateau en mode non autonome ou par les utilisateurs en mode autonome après formation. |
 |
Modalités d'accès
Le plateau peut, sur demande, prendre en charge la préparation des échantillons :
- petites molécules : extraction sur phase solide – SPE ou extraction liquide-liquide avec ou sans ajout de sels QuECHERS
- protéomique : extraction, fractionnement ou enrichissement
Toute demande d’analyses doit être adressée à l’adresse mail contact du pôle Analyses Moléculaires, accompagnée du formulaire adapté téléchargeable ci-dessous.
Pour les utilisateurs autonomes, un calendrier de réservation est mis à disposition.
Pour toute demande ou réservation, un code projet vous sera demandé. Si vous n’en avez pas, merci de remplir le formulaire de dépôt de projet (utilisateurs de l’Université de Limoges uniquement). Pour les utilisateurs extérieurs, merci de prendre contact avec BISCEm pour l’attribution de votre code projet.