![]()
Outline:
The inhibition of arylesterase/paraoxonase (EC 3.1.8.1) by 12 polar organic solvents was studied, using blood serum from healthy donors as the enzyme source. Inhibition curves were described by the Hill equation, with a Hill coefficient close to unity. All solvents were inhibitors. The weakest inhibitor was acetonitrile (C50 ~ 0.35 M), the strongest inhibitor was hexamethylphosphoramide (C50 ~ 0.005 M). The two isozymes (A and B) of arylesterase gave very similar results. Quantitative structure-activity relationships were computed with the molecular lipophilicity potential (MLP) combined with either the experimental parameters of Abraham or the theoretical parameters of Wilson and Famini. The main determinants of inhibition were identified as lipophilicity, molecular volume, and electron-donating ability. Using MLP allowed to take into account the conformation of the inhibitor molecule, giving better results than using the octanol/water partition coefficient (log P). It is suggested that the inhibitor binds to the enzyme by both charge-transfer interactions involving its polar group, and hydrophobic interactions involving its alkyl substituents, as long as these substituents are located at a sufficient distance from the polar group, so that their lipophilicity cannot be masked by the hydrophilicity of the polar group.
Arylesterase (EC 3.1.8.1, also known as paraoxonase) is an enzyme present in the blood of mammals where it is associated with high density lipoproteins (HDL). This enzyme is known mainly for its ability to hydrolyse toxic organophosphorus esters like paraoxon (diethyl-4-nitrophenyl phosphate), the active metabolite of the insecticide parathion. Other synthetic esters such as phenyl acetate are also hydrolysed with a high specific activity. The physiological substrate remains unknown, in spite of the growing body of evidence suggesting the involvement of the enzyme in the pathogenesis of atherosclerosis.1 In previous work, we have shown that the enzyme was inhibited by alcohols.2 The quantitative structure-activity relationships suggested that the alcohols interact with a hydrophobic site consisting primarily of nonpolar aliphatic amino acids. In order to obtain further insight about the inhibiting action of organic solvents on this enzyme, we decided to investigate another series of solvents, more polar than the alcohols, so that they could interact with more hydrophilic amino acids. As previously used with the alcohols, these solvents were characterized by the experimental solvation parameters of Abraham et al,3 the theoretical parameters of Wilson and Famini,4 and several lipophilicity parameters, including the molecular lipophilicity potential.5 The inhibition was studied on the two isoforms (A and B) of the enzyme, using human serum as the enzyme source, and the inhibition parameters were related to the physical properties of the solvents by linear regression analysis.
The physicochemical properties of the solvents were characterized with the experimental solvation parameters, according to the general solvation equation of Abraham:3
![]()
In this equation, SP is some property of a series of compounds in a given system, and the explanatory variables (or descriptors) are compound properties as follows:
| R2 | Excess molar refraction that can be determined simply from a knowledge of the compound refractive index.6 The R2 descriptor represents the tendency of a compound to interact with a phase through pi or n electron pairs (unit = mL.mol-1/10) |
| Compound dipolarity / polarizability,7 it being not possible to devise descriptors for these properties separately. | |
| Overall hydrogen-bond acidity.8 | |
| Overall hydrogen-bond basicity.8 | |
| Vx | McGowan characteristic volume that can be calculated for any compound simply from molecular structure, using a table of atomic constants and the number of bonds, whatever their multiplicity.9 (unit = mL.mol-1/100) |
The coefficients (r, s, a, b, v) in the Abraham equation are found by multiple linear regression analysis. They serve to characterize the differences between the phases in action: in the present case between the receptor site of the enzyme and water. Their significances are as follows : the r-constant is a measure of the difference in propensity of the phases to interact with solute pi or n electron pairs; the s-constant descripts the difference in the phases dipolarity/polarizability; the a-constant is a measure of the phases difference in hydrogen-bond basicity; the b-constant is a measure of the phases difference in hydrogen-bond acidity; and the v-constant is a measure of the difference between the hydrophobicity or lipophilicity of the two phases.
The values of these parameters for the series of 12 organic solvents are presented in Table 1. The structures of the compounds are displayed on Figure 1.
The least polar solvent was ethyl acetate, the more polar being dimethylsulfoxide. The hydrogen bond basicity scale ranged from 0.31 for nitromethane to 1.60 for hexamethylphosphoramide, which is one of the most basic solvents available. The hydrogen bond acidities of the compounds were negligible. All solvents were more polar and less acidic than the alcohols used in our previous study,2 and 7 solvents were more basic than the alcohols.
The theoretical parameters were defined according to the general equation of Wilson and Famini:4, 10
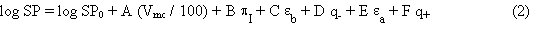
In this equation, SP is the property to be explained, and the explanatory variables are computed from molecular geometries optimized with the MNDO semi-empirical molecular orbital method. These variables are:
| Vmc | Molecular volume (Å3) computed by a Monte-Carlo method. |
| Polarizability index, equal to the polarizability volume divided by Vmc | |
| Hydrogen bond basicity parameter, defined by:
|
|
| q- | Absolute value of the lowest negative charge on the molecule. |
| Hydrogen bond acidity parameter, defined by:
|
|
| q+ | Highest positive charge on the hydrogen atoms of the molecule. |
The coefficients (A, B, C, D, E, F) of the Wilson-Famini equation have significances comparable to those of the Abraham equation. However, in this theoretical model the hydrogen bond acidity or basicity is split into an electrostatic effect (modeled by q+ or q-) and a charge-transfer effect (modeled by ![]() or
or ![]() ). The values of these parameters are presented in Table 2.
). The values of these parameters are presented in Table 2.
The lipophilicity of the solvents was characterized by the logarithm of the octanol/water partition coefficient, log P, for which we used:
![]()
In addition, we used the molecular lipophilicity potential (MLP), which takes into account the conformation of the molecule. The sums of the positive (MLP+, lipophilic) and negative (MLP-, hydrophilic) potentials were divided by 1000 to obtain values in the same range as log P.
These parameters are presented in Table 3. An example of MLP is shown in Figure 2 in the case of dimethylacetamide: it may be seen that the hydrogens on the methyl groups become more lipophilic when their distance from the hydrophilic oxygen increases.
Arylesterase has two isozymes A and B1 which define three phenotypes : A, AB and B (respective genotypes AA, AB and BB). These isozymes differ by their hydrolytic activity towards paraoxon, the B isozyme being the most active. This property allowed the determination of the phenotype of each subject, according to the method of Eckerson et al,11 by computing the ratio of the hydrolysis rates of paraoxon (diethyl-4-nitrophenyl-phosphate) and phenyl acetate. Sera from subjects having the same homozygous phenotype (A or B) were pooled and used as the enzyme sources. All dose-response curves were successfully described by the Hill equation:
v0 = Vmax / [1 + (C50 / C)n]
Since it has been shown that the inhibition of arylesterase is highly dependent on the lipophilicity of the inhibitor,2 the first step in constructing a structure-activity relationship consisted of selecting the best lipophilicity parameter. The pC50 was therefore expressed as a function of each of the five parameters: log P (observed), log P (estimated by Leo's method), log P (estimated by Abraham's method), MLP+ (sum of the positive, or lipophilic, lipophilicity potentials) and MLP- (sum of the negative, or hydrophilic, lipophilicity potentials). The best correlation was obtained with MLP+ (for which r > 0.63, while |r| < 0.42 for the other lipophilicity parameters). In the second step, the selected lipophilicity parameter (MLP+) was combined with either the solvation parameters
(R2 ,
![]() ,
,
![]() ,
,
![]() ,
Vx)
or the theoretical parameters
(Vmc ,
,
Vx)
or the theoretical parameters
(Vmc ,
![]() ,
,
![]() ,
q- ,
,
q- ,
![]() ,
q+) :
the whole regression equation was computed first, then the parameters associated to the least significant regression coefficients were progressively deleted, until the best equation was obtained. The resulting equations are presented in Table 5.
,
q+) :
the whole regression equation was computed first, then the parameters associated to the least significant regression coefficients were progressively deleted, until the best equation was obtained. The resulting equations are presented in Table 5.
The Wilson-Famini model identified MLP+, Vmc , and
![]() as the explanatory variables. These factors explained about 90% of the experimental variance of pC50 for the A and B isozymes. The amount of correlation between the explanatory variables was acceptable (|r| < 0.35). The regression coefficients were significant at the 5% level, except for the coefficient of
as the explanatory variables. These factors explained about 90% of the experimental variance of pC50 for the A and B isozymes. The amount of correlation between the explanatory variables was acceptable (|r| < 0.35). The regression coefficients were significant at the 5% level, except for the coefficient of ![]() with the B isozyme, which was significant only at the 10% level. The comparison of the observed and estimated values of pC50 for the A isozyme is shown in Figure 4. The points were evenly distributed around the theoretical line.
with the B isozyme, which was significant only at the 10% level. The comparison of the observed and estimated values of pC50 for the A isozyme is shown in Figure 4. The points were evenly distributed around the theoretical line.
The Abraham model identified MLP+ and either Vx or
![]() as the explanatory variables. However, combining the 3 parameters into a single equation resulted in non-significant regression coefficients for Vx and
as the explanatory variables. However, combining the 3 parameters into a single equation resulted in non-significant regression coefficients for Vx and
![]() ,
which was due to the high correlation between these parameters (r = 0.92). Hence, only the equations containing either Vx and
,
which was due to the high correlation between these parameters (r = 0.92). Hence, only the equations containing either Vx and
![]() were reported. The correlations between explanatory variables in these equations were acceptable (|r| < 0.11), but of course this procedure resulted in a slight decrease in predictive power with respect to the Wilson-Famini model.
were reported. The correlations between explanatory variables in these equations were acceptable (|r| < 0.11), but of course this procedure resulted in a slight decrease in predictive power with respect to the Wilson-Famini model.
The results obtained with the Wilson-Famini equation suggest that a solvent can interact with arylesterase through hydrophobic interactions and charge-transfer interactions. The hydrophobic interaction is best modeled by the molecular lipophilicity potential (MLP), which takes into account the conformation of the inhibitor, in the sense that, for instance, an hydrophobic group such as CH3 will become less hydrophobic if it is located near an hydrophilic group such as a carbonyl. The hydrophobic interactions are also increased by the molecular volume of the inhibitor. In fact, the MLP, which is derived from the octanol-water partition coefficient, incorporates a volume effect (via Eq. 3). The existence of an additional volumic term in the structure-activity equation suggests that the transfer of a molecule from the aqueous environment into the enzyme binding site occurs more easily than its transfer into octanol. This is in agreement with the results of our previous analysis with alcohols (ref.2 and unpublished results), from which we can compute a binding free energy of 1.4 kcal/mol for each CH2 group, which is about twice the transfer free energy from water to octanol (0.69 kcal/mol, estimated from Hansch's parameter pi = 0.5). Similar results have been observed for other enzymes.12 These findings, which mean that the enzyme site is more hydrophobic than octanol, are usually interpreted by assuming that a cavity preexists on the enzyme surface, while the creation of such a cavity in octanol requires the breaking of some intermolecular bonds.
The charge-transfer interactions were best modeled by the electronic parameter ![]() which characterizes the electron-donating ability of the inhibitor. In the original model of Wilson and Famini this parameter is related to hydrogen bond basicity, in association with the most negative charge q- on the molecule. However, the absence of q- in the final regression equation suggests that the binding of the inhibitor does not involve an hydrogen bond. The electron acceptor is probably an aromatic amino acid. Although the three-dimensional structure of the enzyme is unknown, it has been shown by directed mutagenesis that several histidine and tryptophan residues are required for the catalytic activity.
13,
14,
15
Another possible electron acceptor could be the calcium ion which is bound to the active site and serves as an activator. However, in such a case one would expect an electrostatic interaction, and consequently the presence of q- in the equation.
which characterizes the electron-donating ability of the inhibitor. In the original model of Wilson and Famini this parameter is related to hydrogen bond basicity, in association with the most negative charge q- on the molecule. However, the absence of q- in the final regression equation suggests that the binding of the inhibitor does not involve an hydrogen bond. The electron acceptor is probably an aromatic amino acid. Although the three-dimensional structure of the enzyme is unknown, it has been shown by directed mutagenesis that several histidine and tryptophan residues are required for the catalytic activity.
13,
14,
15
Another possible electron acceptor could be the calcium ion which is bound to the active site and serves as an activator. However, in such a case one would expect an electrostatic interaction, and consequently the presence of q- in the equation.
The Abraham model identified the same explanatory variables, but could not discriminate the effects of molecular volume and hydrogen bond basicity, since these two parameters were highly correlated.
In addition to binding to the enzyme, the organic solvent may act by modifying the physico-chemical properties of the medium. The substrate partitions between the enzyme and the water-cosolvent mixture. A stronger interaction of the substrate with the medium would result in an apparent inhibition. In order to get a structure-activity relationship similar to the observed one, the water-cosolvent mixture should become more acidic and more lipophilic. The first possibility may be ruled out since all organic solvents were less acidic than water. On the other hand, an increased lipophilicity of the water-cosolvent mixture has already been observed, e. g. with the solvent systems used in liquid chromatography.16 However, such modifications occured at relatively high concentrations of the cosolvent, and were adequately described by its molecular volume, without need for a conformation-dependent parameter such as the lipophilicity potential. So, we consider that the specific binding of the organic solvent to the enzyme was the predominant phenomenon in our study. The importance of hydrophobic interactions in enzyme systems is widely recognized, and the involvement of charge-transfer interactions has been found in other esterases17.
This study showed that a polar solvent can interact with arylesterase both by the electron-donating ability of its polar group (e.g. carbonyl, phosphoryl...) and by the lipophilicity of its alkyl substituents, provided that these substituents are located at a sufficient distance from the polar group. This conformational effect was accounted for by the molecular lipophilicity potential, which was combined with the Abraham or Wilson-Famini parameters in order to get an efficient quantitative structure-activity relationship. This approach seems promising and could be extended to other biological processes, since the molecular lipophilicity potential is easily estimated from molecular models.
All organic solvents were of commercial origin and at the highest available purity (> 99%). Since no purified preparation of arylesterase was available to us at the time of this study, sera from healthy donors were used as the enzyme source. Arylesterase activity was determined by the method of Eckerson et al,11 using phenyl acetate as substrate. The reaction mixture contained 5 �L of serum in 1.5 mL of Tris/HCl buffer (9 mM, pH 8.0 at 25�C, containing 0.9 mM CaCl2) and 0.5 mL of an inhibitor solution in buffer. This mixture was preincubated at 25�C for 2 min, then the reaction was started by adding 0.5 mL of a substrate solution (phenyl acetate, 5 mM) in buffer. The substrate concentration in the reaction mixture was 1 mM. No cosolvent was used to dissolve the phenyl acetate, which was quite soluble in water at the concentration used. The absorbance rise due to the production of phenol was measured at 25�C and 270 nm (molar extinction coefficient: 1310 L.mol-1.cm-1) in a Shimadzu UV 1205 spectrophotometer equipped with a Peltier cooling unit. Absorbances were measured every 0.5 s for 30 s, then transferred to a microcomputer by means of a RS-232 interface. The initial reaction rate was estimated by fitting a polynomial to the kinetic curve. The Hill equation was fitted to the experimental data by unweighted nonlinear regression, using our computer program WinReg.18
All computations were performed on a PC computer. Theoretical parameters were computed with MOPAC 6.019 using geometries optimized with the MNDO semi-empirical molecular orbital method. Molecular volumes were estimated by ARVOMOL.20 Experimental values of log P were taken from the database of the CLOGP 4.0 software,21 which was also used to compute the estimated values according to Leo's method.22 The molecular lipophilicity potential was computed with CHIME,23 using a modification of a program written by Martz.24, 25 Linear regression analysis was performed by WinReg.18
This work was supported in part by grants from the French Ministère de la Santé (Programme Hospitalier de Recherche Clinique, 2001) and from the Fondation pour la Recherche Médicale. The expert technical assistance of Jean-Herv� Comte is gratefully acknowledged.