Diagnostics moléculaires de neuropathies d'origine génétique
Les patients atteints de neuropathie suivis au CHU de Limoges peuvent être diagnostiqués par méthodes moléculaires (Séquençage Nouvelle Génération – NGS) au sein du Service de Biochimie et Génétique Moléculaire du CHU de Limoges, grâce à la plateforme de Séquençage Genolim de l’UMS BISCEM et de l’UF de Bioinformatique médicale qui a vu le jour au sein du Centre Hospitalier.
Variants de structure (SVs)
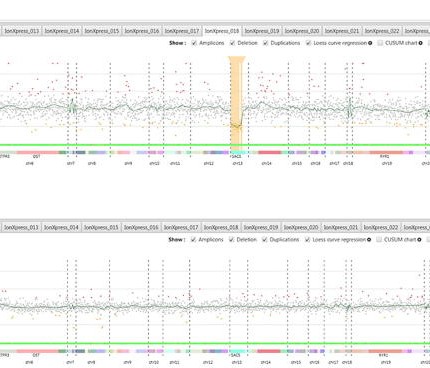
Une attention toute particulière a été portée sur l’identification et la caractérisation de Variants de Structure (SVs) pathogènes responsables de la maladie de Charcot-Marie-Tooth (CMT), puisqu’environ 60% des patients n’ont pas de cause moléculaire identifiée pour leur pathologie.
A ce jour, plus de 120 gènes sont établis comme pouvant être responsables de la maladie de CMT, mais une forte proportion de patients testés ne sont pas diagnostiqués avec des mutations connues ou probables de ces gènes. Cependant, quelques Variants de Structure, différents de la duplication de PMP22 ont été rapportés.
L’un des axes de recherche de l’unité est donc d’identifier ces nouveaux SVs. Ainsi, des outils bio-informatiques permettant de détecter des SVs à partir de données NGS ont été développés : logiciels Cov’Cop et CovCopCan. Ils sont publiés et disponibles gratuitement.
Les résultats préliminaires, obtenus à partir de ces logiciels, suggèrent qu’il existe plusieurs SVs pathogènes responsables de la maladie de CMT. Les SVs recherchés seront des délétions, des duplications, mais également d’éventuelles mosaïques à partir des données NGS. Par la suite, il est prévu d’étendre cette exploration aux données obtenues à partir d’exomes.
Grâce à ces approches, de nouveaux SVs pourront être identifiés dans les gènes d’intérêt ainsi que dans d’autres non reconnus à ce jour, mais la caractérisation de ces types de mutations actuellement sous diagnostiquées et donc sous-estimées sera possible. Il a été établi qu’un tiers des patients qui présentent les signes cliniques de CMT, ne présentent pas les altérations génétiques qui sont actuellement décrites dans la littérature et reconnues pour induire la maladie.
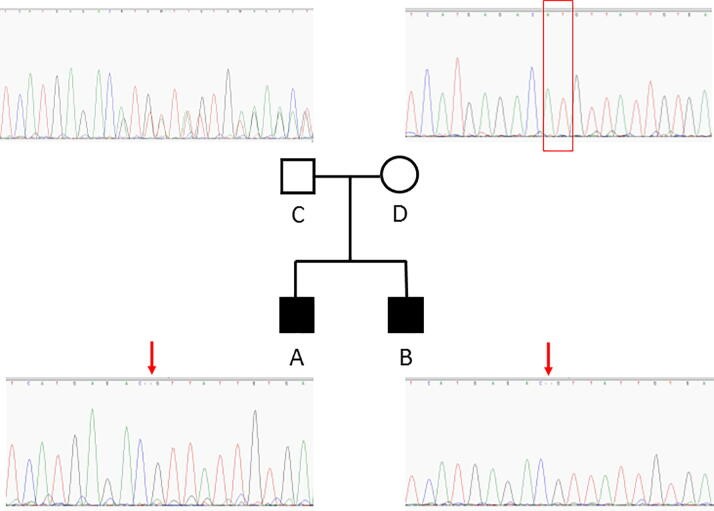
L’identification de nouveaux gènes est un point important pour mieux comprendre la physiopathogénicité des CMT, mais aussi pour ne pas laisser les patients dans l’errance et l’absence de diagnostic.
Mutants d'épissage
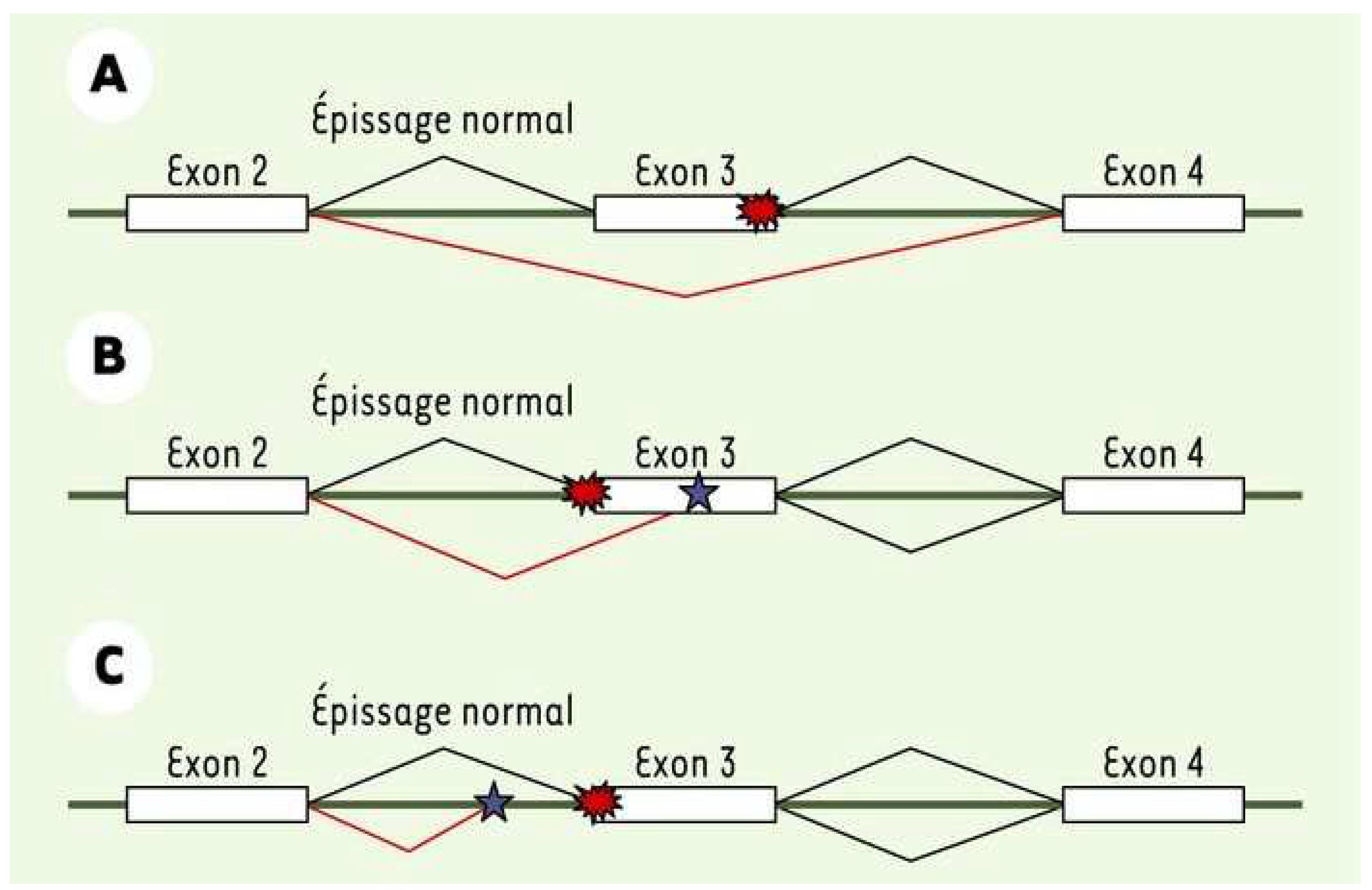
Une seconde attention a été portée sur les potentiels mutants d’épissage qui peuvent être observés dans les gènes impliqués dans la maladie de Charcot-Marie-Tooth (CMT) et les neuropathies périphériques. Nombreux sont prédits comme pouvant induire des anomalies d’épissage par différents logiciels de prédiction. Ainsi, ces erreurs engendreraient un ARNm non conformes et donc une protéine modifiée.
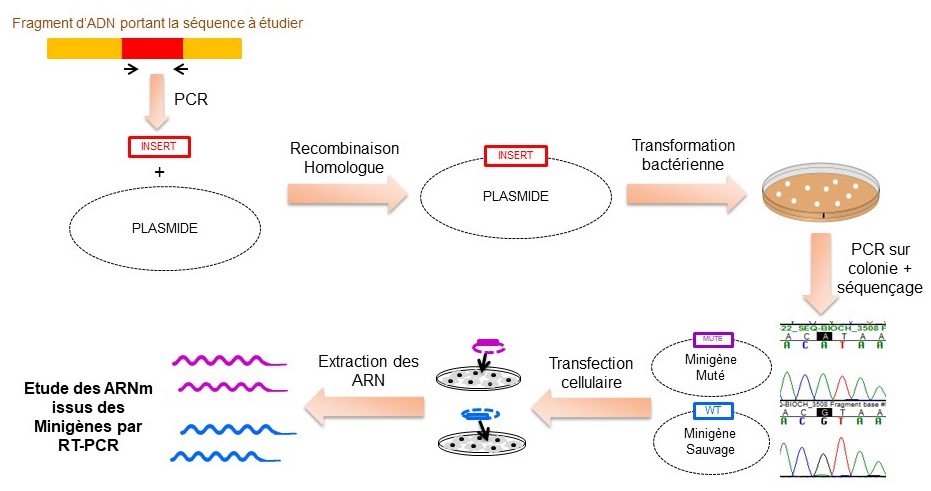
Afin de confirmer la potentielle pathogénicité de ces mutations, le projet « Minigène » a été mis en place pour étudier les effets de ces altérations sur l’épissage de l’ARNm en les créant in vitro. Cette étude se déroule en 2 étapes : (i) la création des « Minigènes » contenant le fragment d’intérêt (sauvage et muté) par différentes techniques de biologie moléculaire, (ii) l’étude de l’effet des mutations sur l’épissage des ARNm du Minigène ainsi obtenu grâce à son utilisation en transfection cellulaire avec différentes lignées cellulaires. L’études des ARNm par RT-PCR sera effectuée afin de comparer les profils d’épissage du Minigène sauvage et du Minigène muté.
Quelques articles représentatifs
– Functional Evaluation of Splice Variants Using a Minigene Strategy. Nizou et al. Methods Mol Biol. 2025:2962:155-170. PMID: 40699428
– The First Large Deletion of ATL3 Identified in a Patient Presenting with a Sensory Polyleuropathy. Pyromali et al. Biomedicines. 2023 May 28;11(6):1565. PMID: 37371660
– From Negative to Positive Diagnosis: Strucutral Variation Could Be the Second Mutation You Are Looking for in a Recessive Autosomal Gene. Pyromali et al. J Pers Med. 2022 Feb 3;12(2):212. PMID: 35207700
– New structural variations responsible for Charcot-Marie-Tooth disease: The first two large KIF5A deletions detected by CovCopCan software. Pyromali et al. Comput Struct Biotechnol J. 2021 Jul 30;19:4565-4272. PMID: 34429846
– One multilocus genomic variation is responsible for a severe Charcot-Marie-Tooth axonal form. Miressi et al. Brain Sci. 2020 Dec 15;10(12):986. PMID: 33333791
– A mutation can hide another one: Think Structural Variants! Miressi et al. Comput Struct Biotechnol J. 2020 Aug 2;18:2095-2099. PMID: 32832037
– CovCopCan: An efficient tool to detect Copy Number Variation from amplicon sequencing data in inherited diseases and cancer. Derouault et al. PloS Comput Biol. 2020 Feb 12;16(2):e1007503. PMID: 32049956
– New PRPS1 variant p.(Met68Leu) located in the dimerization area identified in a French CMTX5 patient. Lerat et al. Mol Genet Genomic Med. 2019 Sep;7(9):e875. PMID: 31338985
– COV’COP allows to detect CNVs responsible for inherited diseases among amplicons sequencing data. Derouault et al. 2017 May 15;33(10):1586-1588. PMID: 28137711