ANNEXES
1. Porosimétrie au mercure
La porosimétrie au mercure est largement utilisée pour caractériser la porosité de matériaux pulvérulents ou compacts. Par pénétration de mercure sous pression, on peut déterminer la taille et la quantité des espaces vides et des pores des matériaux et leur densité. Le mercure expulsé des pores lorsque la pression appliquée décroît donne des informations sur la taille et la structure des pores.
Chaque fois qu’un liquide présente un angle de mouillage supérieur à 90° avec un solide, il mouille difficilement celui-ci et ne pénètre dans les pores que s’il est comprimé. Le mercure présente, avec un grand nombre de matériaux, un angle de contact supérieur à celui d’autres liquides usuels. Cet angle varie de 112 à 142°, 130° étant la valeur la plus fréquemment rencontrée.
La porosité est modélisée sous forme de cylindres de section circulaire et de rayon r. La tension de surface du mercure s’opposant à son infiltration,il faut appliquer une pression externe P. A l’équilibre, la relation de Washburn relie la pression au rayon des pores infiltrés.
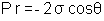
avec P la pression appliquée sur le fluide
r le rayon du pore à infliltrer
σ la tension de surface du liquide sur la surface du pore
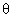 l’angle de mouillage du fluide sur le matériau
l’angle de mouillage du fluide sur le matériau
Cette équation montre qu’il n’y a aucune pénétration pour une pression nulle. Quand la pression augmente, le volume de mercure qui pénètre traduit un volume de pores dont la taille correspond à la pression instantanée. Les résultats sont fournis sous forme d’une courbe volume des pores/taille des pores.
Cette méthode est sensible pour des pores de faible diamètre (0,003.10-6 à 10.10-6 m), mais limitée dans le domaine des pores de grande taille (d>150.10-6 m). Les pores très larges (d=300.10-6 m), se remplissant en même temps que la cellule, ne peuvent pas être comptabilisés à la pression initiale.
2. Surface spécifique (méthode BET)
Lorsqu’un solide est exposé à un gaz, les molécules de gaz s’adsorbent sur sa surface. La mesure de la quantité de gaz adsorbée permet de déterminer la surface spécifique du solide. La variation de la quantité de gaz adsorbée à température constante en fonction de la pression est appelée isotherme d’adsorption. L’exploitation mathématique de cette courbe a été réalisée par Brunauer, Emmett et Teller avec les hypothèses suivantes:
L’adsorption est localisée sur des sites bien définis, chacun d’eux n’admettant qu’une seule molécule adsorbée ; tous les sites possèdent la même énergie et les molécules adsorbées n’ont pas d’interactions entre elles
L’adsorption s’effectue dès le début en plusieurs couches, les molécules adsorbées dans la première couche servant de sites d’adsorption pour les molécules de la deuxième couche et ainsi de suite.
Il existe un équilibre permanent entre les molécules qui s’adsorbent à la surface et le nombre de molécules qui s’en désorbent. La désorption est un processus activé dont l’énergie d’activation est E1 pour la première couche adsorbée et EL pour les couches suivantes.
Le traitement mathématique de ces hypothèses permet de mettre la relation sous la forme suivante :
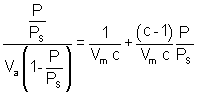
avec P la pression d’équilibre
PS la pression de vapeur saturante du gaz à la température d’adsorption
Va le volume moléculaire adsorbés à la pression P
Vm le volume correspondant à la formation d’une monocouche
c une constante caractéristique du système gaz-solide étudié
Cinq types d’isothermes d’adsorption découlent de la formule ci dessus. Cependant, la concordance entre les isothermes théoriques et expérimentales est généralement limitée à certain domaine de pression. Cette situation résulte du modèle choisi par Brunauer, Emmett et Teller. En effet, ce modèle ne tient pas compte de l’hétérogénéité de la surface du solide qui se manifeste pour les faibles pressions, ni des interactions latérales entre les molécules adsorbées qui modifient l’isotherme lorsque la pression atteint une valeur suffisante. L’équation BET est en général utilisée pour des pressions relatives comprises entre 0.05 et 0.35.
La valeur de Vm permet de calculer la surface spécifique. En effet, Vm est le volume de vapeur nécessaire pour recouvrir la surface d’un gramme de solide d’une couche monomoléculaire, la valeur de la surface spécifique est égale à la surface occupée par une molécule de vapeur multipliée par le nombre de molécules contenues dans Vm.
3. Spectrométrie Auger
L’effet Auger est la désexcitation, d’un atome ionisé, par un processus non radiatif. Le trou, formé par l’ionisation d’un atome dans une couche basse de son nuage électronique, est rapidement comblé par un électron d’une couche supérieure. L’énergie dissipée peut prendre plusieurs formes, un rayonnement X, utilisé en microsonde X ou fluorescence X, ou être transférée à un autre électron qui est éjecté de l‘atome avec une énergie EA déterminée par les trois niveaux d’énergie mis en jeu.
Les spectres d’électrons Auger sont l’addition de deux composantes principales, un fond continu d’électrons rétrodiffusés et des pics correspondant aux ionisations des atomes présents.
L’essentiel du spectre des électrons secondaires, des électrons rétrodiffusés et des électrons secondaires de basse énergie peut être utilisé pour former une image de la surface. Ils possèdent chacun une information différente.
Dans un microscope électronique à balayage conventionnel, le faible niveau de vide entraîne une forte contamination de la surface qui modifie les variations d’émission dues à la composition. Le contraste de l’image est donc largement déterminé par la topographie de l’échantillon. Avec les appareils utilisés pour les mesures Auger, qui travaillent sous ultra vide, le contraste de l’image est bien meilleur.
Les électrons primaires sont moins sensibles aux contaminants de surface, mais dépendent beaucoup du numéro atomique de l’élément constitutif de l’échantillon. Le coefficient de rétrodiffusion dépend fortement du numéro atomique de l’élément mais est quasi indépendant de l’énergie du faisceau initial. Les images formées à partir de ces électrons permettent de différencier les éléments.
4. Microanalyse X
La microanalyse X par sonde électronique permet l’analyse qualitative et quantitative dans un très petit volume de matière, de l’ordre du 10-18 m3. La limite de détection est d’environ 10-17 à 10-18 kg, ce qui correspond à une teneur inférieure à 100ppm. Elle permet l’analyse de tous les éléments de nombre atomique supérieur à 4 (béryllium).
La résolution obtenue en microanalyse est liée à la dimension du volume analysé, qui dépend de la tension d’accélération, de la cible et de l’énergie d’ionisation du niveau considéré. Cette résolution est actuellement de l’ordre du micromètre. Toute phase ou tout mélange de phases de dimensions inférieures à cette résolution est difficile sinon impossible à analyser. Il en est de même pour l’étude des concentrations aux limites de phases. L’existence d’un fond continu, dans le spectre de rayons X, représente une limite pour la détection d’éléments en faible trace. Cette limite est de l’ordre de 40 à 100ppm en WDS et de l’ordre de 0,05 à 0,1% en EDS.