CHAPITRE 3 : COMPORTEMENT A L’OXYDATION DES MATERIAUX HfB2-SiC
La résistance à l’oxydation de composites massifs à base de borures et de carbures réfractaires, le plus souvent élaborés par frittage sous charge, a été étudiée depuis de nombreuses années. Les systèmes Zr/B/Si/C et Hf/B/Si/C ont fait l’objet de plusieurs travaux dans le cadre d’applications à très hautes températures, comme les corps de rentrée atmosphérique(3, 4, 5, 6, 7, 13, 15).
Après avoir présenté les comportements à l’oxydation de ZrB2, HfB2 et SiC, nous montrerons que les interactions entre les produits de la réaction au cours de la corrosion de composites ZrB2-SiC et HfB2-SiC leur procurent des propriétés intéressantes dans une large gamme de température et de pression d’oxygène. Nous décrirons ensuite les différents mécanismes susceptibles d’intervenir lors de l’oxydation de substrats carbonés protégés par une barrière de diffusion. Enfin nous expliciterons les cinétiques d’oxydation en fonction de la température et de la pression partielle d’oxygène des matériaux élaborés par infiltration réactive. Les résultats seront discutés en relation avec la microstructure des matériaux et des produits de réactions.
1. RAPPELS BIBLIOGRAPHIQUES
1.1 Oxydation de MeB2 (Me=Hf, Zr)
Les diborures de métaux s’oxydent selon la réaction générale suivante :
MeB2 (s) + 5/2 O2 (g) → MeO2 (s) + B2O3 (l ou g) [1]
L’étude de la cinétique d’oxydation présente une difficulté majeure dans la mesure où B2O3 est très volatil au-delà de 1373K. Deux domaines de température doivent donc être distingués en fonction des espèces formées et le simple suivi de la variation de masse de l’échantillon n’est pas suffisant pour déterminer la cinétique de la réaction.
Pour des températures inférieures à 1373K, il se forme une couche de MeO2-B2O3 protectrice et la variation de masse totale a une allure parabolique (19, 14, 2). L’oxydation est alors limitée par la diffusion de l’oxygène à travers cette barrière. Au-dessus de 1373K, la consommation d’oxygène est plus rapide à cause de la diminution de l’épaisseur de la couche de B2O3 et la cinétique d’oxydation s’accélére. Deux domaines d’oxydation ont été mis en évidence sur HfB2(2, 14). Ce phénomène est indépendant de la présence de B2O3 car, au-dessus de 1473K, sa vitesse de formation est égale à sa vitesse de volatilisation. Cette discontinuité est associée à la transformation allotropique de HfO2, à 1973K. Dans le cas de ZrB2, la transition de phase intervient à 1373K. Il est difficile d’isoler son influence par rapport au départ de B2O3.
Selon Kaufman et al(14) la cinétique d’oxydation de HfB2 est parabolique jusqu’à 2300K, mais l’influence de la pression partielle d’oxygène varie selon les auteurs. Kaufman et al(14) ont mesuré des vitesses d’oxydation indépendantes de la pression partielle d’oxygène, entre 5.103 et 2.104 Pa, à 2300K, alors que Berkowitz et al(2) ont trouvé un exposant de la loi de pression d’oxygène de 0,4±0,1 entre 1,33.102 et 2,66.104Pa à 1630K et 1760K. Cette différence de comportement peut être attribuée à la morphologie de l’oxyde formé. L’oxydation de HfB2 au-dessus de 2000K conduit à une couche d’oxyde colonnaire alors qu’en dessous, les grains sont équiaxes. La vitesse d’oxydation pourrait être différente selon l’orientation cristalline de l’oxyde(14).
Kaufman et al(14) ont étudié la résistance à l’oxydation de HfB2 et ZrB2 en fonction de leur stœchiométrie. Il semble qu’un excès en métal améliore les performances par transformation de B4C, présent sous forme d’impureté, en une phase contenant Hf, B et C. La volatilité de B2O3 pourrait également être plus faible, la présence de métal diminuant l’activité du bore.
En conclusion, les études d’oxydation ont permis de classer les diborures par ordre croissant de résistance à l’oxydation(14, 5, 6, 7) :
HfB2 > ZrB2 > TiB2 > TaB2 > NbB2
La meilleure tenue de HfB2 et ZrB2 s’explique par la formation d’oxydes ultra-réfractaires.
1.2 Oxydation de SiC
Les travaux sur l’oxydation du carbure de silicium ont révélé l’existence de deux régimes cinétiques. Dans le domaine d’oxydation passive, sous de fortes pressions partielles d’oxygène, il se forme une couche protectrice de silice selon l’équation [2] qui induit un régime diffusionnel.
SiC(s) + 3/2 O2(g) → SiO2(s) + CO(g) [2]
A haute température et sous faible pression d’oxygène, l’oxydation active [3] résulte du départ de produits gazeux.
SiC(s) + O2(g) → SiO(g) + CO(g) [3]
Trois équilibres thermodynamiques peuvent être envisagés :
SiC(s) + 2SiO2(s) ⇆ 3 SiO(g) + CO(g)
SiC(s) + SiO2(g) ⇆ 2SiO(g) + C(s)
2 SiC(s) + SiO2(s) ⇆ 3 Si(s) + 2CO(g)
Le premier semble le plus probable car dans la plupart des études, ni carbone, ni silicium n’ont été détectés dans les produits de l’oxydation (1, 11).
Les travaux sur la transition oxydation passive, oxydation active font apparaître une grande dispersion des résultats. Balat(1) a réalisé une analyse des résultats publiés et une étude comparative sur du carbure de silicium fritté et du carbure de silicium obtenu par CVD. Il ressort de cette expérimentation que pour une température donnée, la transition entre les deux domaines dépend de la microstructure du matériau et des conditions expérimentales : pression partielle d’oxygène, pression gazeuse totale, débit des gaz…(8, 17, 18, 21). De plus, la présence d’oxygène dissocié élargit le domaine d’oxydation passive(1).
En ce qui concerne l’oxydation passive du carbure de silicium, deux domaines ont été mis en évidence. Pour des températures comprises entre 1273K et 1573K, les cinétiques d’oxydation en régime isotherme suivent une loi parabolique. Un domaine linéaire est observé aux tous premiers instants de la réaction, pour de très faibles épaisseurs de silice. Les valeurs de l’énergie d’activation publiées dans la littérature correspondant au processus limitant sont très dispersées suivant les matériaux étudiés mais les auteurs s’accordent pour l’attribuer à la migration centripéte de l’oxygène moléculaire à travers la silice. L’absence de gradient de carbone dans l’oxyde et les calculs de la vitesse de transport de CO montrent que sa diffusion n’est pas le phénomène limitant. De plus, on observe une bonne adéquation entre les valeurs des constantes de diffusion et celles calculées à partir de la perméation de l’oxygène à travers la silice amorphe.
A plus haute température (1573K-1773K), le deuxième régime de diffusion observé correspond à une migration, en parallèle, d’oxygène moléculaire et d’oxygène ionique. La diffusion ionique devient prépondérante quand la température augmente. L’étude de l’influence de la pression d’oxygène montre que les constantes de diffusion Kp sont proportionnelles à cette dernière dans le premier domaine, conformément à la diffusion de l’oxygène moléculaire, alors qu’au dessus de 1623K, Kp varie selon PO2 0,7 au lieu de PO2 0,5 pour une diffusion d’oxygène ionique. Cette valeur intermédiaire de l’exposant confirme l’existence des deux espèces diffusantes.
1.3 Oxydation du composite (Zr)HfB2-SiC
Dans le but d’améliorer la tenue de HfB2, des composites HfB2/SiC ont été élaborés par frittage sous charge(4, 5, 6, 7, 20). Les cinétiques d’oxydation en régime isotherme sont paraboliques dans une large gamme de température mais deux domaines sont mis en évidence. Jusqu’à 1623K, la vitesse d’oxydation du composite est proche de celle du diborure car ce dernier s’oxyde préférentiellement. A partir de 1623K, la cinétique se ralentit fortement car le carbure de silicium commence à s’oxyder et il se forme un borosilicate qui peut être protecteur selon les conditions de température et de pression. Des particules de SiC se retrouvent dans la couche oxydée jusqu’à 1673K sous une pression partielle d’oxygène de 3,33.104Pa(12).
Les produits de la réaction d’oxydation dépendent de la température et de la pression partielle d’oxygène. MeO2 (avec Me=Zr, Hf) est observé dans tous les domaines de température (1073-2273K) et pour des pressions partielles d’oxygène de 0,3.102 à 3,33.104 Pa. B2O3 sous forme liquide apparaît en dessous de 1373K pour une pression partielle d’oxygène supérieure à 10 Pa, mais se volatilise rapidement passée cette température. Cependant, en présence de silice, il se forme un verre borosilicaté qui s’appauvrit progressivement en B2O3 à plus haute température. La silice provient de l’oxydation du carbure de silicium, dans le domaine d’oxydation passif. A très haute température et faible pression partielle d’oxygène, la silice devient instable et SiO(g) se dégage.(20, 9)
Pour les matériaux frittés sous charge dont les grains ont un diamètre de plusieurs microns, le comportement à l’oxydation, en dessous de 1373K est donc proche de celui de ZrB2 ou HfB2, jusqu’au départ de B2O3 et au début de l’oxydation de SiC.
Le comportement à haute température est essentiellement lié à la présence de silice qui induit une étape limitante de diffusion. Aucune interaction entre ZrO2 ou HfO2 et la phase vitreuse n’a été mise en évidence. Seule la formation de HfO2 en faible quantité a été observée.
2. MECANISMES D’OXYDATION DE MATERIAUX CARBONES PROTEGES PAR UNE BARRIERE DE DIFFUSION A L’OXYGENE
Luthra(16) a publié une étude théorique de l’oxydation des matériaux carbonés protégés par une barrière de diffusion à l’oxygène. Les différents mécanismes limitants sont décrits, en particulier la diffusion de l’oxygène à travers B2O3 liquide.
Les étapes du mécanisme d’oxydation sont les suivantes (figure 55).
- Diffusion du gaz oxydant à travers la couche limite
- Diffusion à travers les fissures si la barrière de diffusion est fissurée. Pour une taille des fissures importante, la diffusion est rapide. En revanche lorsque la taille des fissures est faible, la diffusion s'effectue par effet Knudsen.
- Diffusion en volume qui dépend des défauts présents dans le matériau.
- Réaction à l'interface revêtement/substrat.
Figure 55 : Mécanismes d’oxydation du carbone à travers une barrière de diffusion à l’oxygène(16)
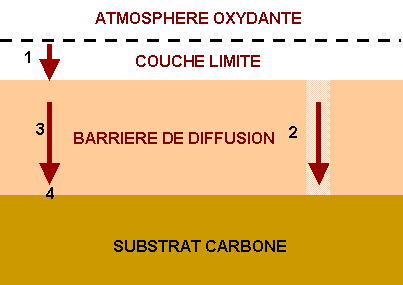
Chacune des étapes du processus d'oxydation peut être limitante et déterminer la cinétique d'oxydation globale du substrat carboné.
L’étude est basée autour de quatre phénomènes correspondant aux situations rencontrées lors de l’utilisation, la diffusion en phase gazeuse dans les fissures, la diffusion à travers un film dense et continu de B2O3, la diffusion dans des fissures remplies de B2O3 liquide et l’influence du CO formé à l’interface entre la protection et le substrat.
Les calculs sont comparés aux performances attendues pour deux applications à court et long terme, correspondant respectivement à 72000 s et 7200000 s d’utilisation sous une pression partielle d’oxygène de 105Pa. Cette étude se limite aux basses températures (<1473K) car la vitesse d’évaporation de B2O3 n’est pas prise en compte.
2.1 Diffusion en phase gazeuse dans les pores ou les fissures
Dans la plupart des cas, les revêtements sont fissurés à l’issue de l’élaboration ou se fissurent lors de l’utilisation à cause des contraintes thermiques induites par la différence des coefficients de dilatation thermique entre le substrat et la protection.
La diffusion dans les fissures peut être décomposée en deux termes :
Ces phénomènes se produisent en parallèle, le coefficient de diffusion total dans la fissure, Dc peut être calculé comme suit :
 [4]
[4]
Le coefficient de diffusion en phase gazeuse est défini à partir du flux de gaz et de la loi de Fick :
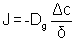 [5]
[5]
avec J le flux de gaz
ΔC la différence de concentration
δ la profondeur de la fissure
Le coefficient de diffusion par effet Knudsen peut être évalué à partir de la relation suivante :
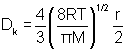 [6]
[6]
avec T la température en Kelvin
r le diamètre des pores
M la masse moléculaire de l'espèce diffusante
La diffusion à travers les pores est plus difficile qu’à travers les fissures car dans le second cas les collisions avec les parois ne se produisent qu'en deux dimensions, le long de la fissure et le libre parcours moyen est plus grand. Plus la taille du défaut diminue, plus la diffusion est difficile et le libre parcours moyen petit.
Le flux d'oxygène le long d'une fissure peut être calculé à partir de la première loi de Fick :
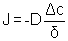 [7]
[7]
avec J le flux de gaz arrivant à la surface du matériau
D le coefficient de diffusion de CO dans l’oxyde (D=Dc)
Δc la différence de concentration de l'espèce gazeuse de part et d'autre du revêtement
δ épaisseur du revêtement
et en multipliant par la concentration de fissures à la surface du film d'oxyde (fc).
avec
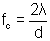 [8]
[8]
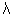 largeur des fissures
largeur des fissures
d l'espace entre deux fissures
On obtient alors le flux moyen d'oxygène à travers tout le revêtement.
2.2 Diffusion à travers un film dense de B2O3 liquide
La diffusion de l’oxygène à travers un film dense peut être calculée à partir de l’expression suivante :
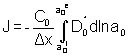 [9]
[9]
avec J le flux d’oxygène à travers la couche dense
C0 la concentration en oxygène dans l’oxyde
a0 l'activité de l'oxygène, les exposants i et e correspondent respectivement aux interfaces internes et externes
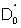 le coefficient de diffusion de l'oxygène
le coefficient de diffusion de l'oxygène
Δx l’épaisseur du film
Le coefficient de diffusion de l’oxygène à travers un film dense peut être relié à la pression d'oxygène par la relation suivante :
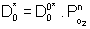 [10]
[10]
avec 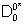 le coefficient de diffusion de l’oxygène à P=1atm
le coefficient de diffusion de l’oxygène à P=1atm
n coefficient qui dépend des défauts de structure.
Le flux d’oxygène à travers le film dense sera alors :
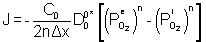 [11]
[11]
avec 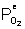 la pression d’oxygène à l’interface externe
la pression d’oxygène à l’interface externe
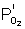 la pression d’oxygène à l’interface interne
la pression d’oxygène à l’interface interne
n coefficient qui dépend des défauts de structure
Δx l’épaisseur du film
C0 la concentration en oxygène dans le matériau
Luthra considére une couche de B2O3 protecteur. L'oxygène diffuse de façon moléculaire entre 624K et 923K. Les défauts majoritaires sont O2,i et son coefficient de diffusion est :
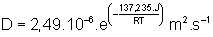 [12]
[12]
En considérant ce type de défauts, l'exposant n dans la relation [10] est égal à 1. Comme l'oxygène diffuse sous forme moléculaire, on a :
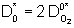 [13]
[13]
L'expression du flux d'oxygène à travers le film de B2O3 est donnée par la relation suivante :
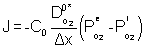 [14]
[14]
avec 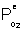 la pression d'oxygène dans la phase gazeuse.
la pression d'oxygène dans la phase gazeuse.
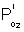 la pression d'oxygène dans le B2O3.
la pression d'oxygène dans le B2O3.
Si la diffusion à travers le B2O3 est limitante 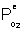 est très supérieure à
est très supérieure à 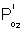 . On peut donc simplifier l'expression précédente.
. On peut donc simplifier l'expression précédente.
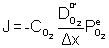 [15]
[15]
On suppose dans ce cas que le CO formé en dessous du film de B2O3 ne le déchire pas. Les calculs montrent que la température maximale d'utilisation d'une protection en B2O3 est 873K et 1123K respectivement pour des applications long terme et court terme (figure 56).
Figure 56 : Vitesses d’oxydation du carbone calculées avec pour étape limitante la diffusion à travers une couche de B2O3.(16)
2.3 Diffusion dans des fissures remplies de B2O3 liquide
La diffusion dans des fissures remplies de B2O3 liquide est très différente de la diffusion dans des fissures ouvertes. Dans ces dernières, les collisions contre les parois influencent la vitesse de transport du gaz pour de faibles diamètres. En présence de liquide, le libre parcours moyen est beaucoup plus petit, les collisions peuvent être négligées et la vitesse d’oxydation à la base de la fissure sera la même que celle calculée pour une couche dense et continue d’oxyde.
La cinétique d’oxydation peut être calculée en multipliant la vitesse maximale d’oxydation [7] par la concentration de fissures [8].
2.4 Effet du monoxyde de carbone
Lors de l'oxydation du carbone, la formation de monoxyde ou de dioxyde de carbone est susceptible de conduire à un décollement du revêtement si la pression devient trop élevée ou à la formation de bulles, si la barrière de diffusion devient visqueuse.
Le tableau 18 montre que les pressions d’équilibre de CO à l’interface entre un substrat carboné et un revêtement à différentes températures peuvent être considérables. Dans les mêmes conditions, les pressions de CO2 seraient encore plus élevées. Par conséquent, elles sont très souvent à l’origine du décollement ou du déchirement de la barrière de diffusion.
| Température (K) | PCO (Pa) |
| 1273 | 1,6.1014 |
| 1473 | 3,6.1013 |
| 1673 | 1,2.1013 |
| 1873 | 4,8.1012 |
Pour que des bulles se forment, il faut que PCO dans le liquide soit inférieure à PCO à l'interface. Celle-ci dépend essentiellement des caractéristiques du film, en particulier, la perméabilité à l'oxygène et au CO. Les calculs montrent que les bulles de CO se forment pour une température supérieure à 1848K et une pression de CO supérieure à 105Pa pour un film de B2O3.
Quatre cas sont envisageables en fonction de l’étape limitante :
- Si la diffusion de l'oxygène à travers la couche d'oxyde est limitante, la pression de CO et la pression d'oxygène seront très inférieures à 105Pa. Il n'y aura pas de bulles. Dans ce cas, la perméabilité de CO à travers le film d'oxyde est très supérieure à celle de l'oxygène. Il n'existe pas d'informations sur la perméabilité du B2O3 au CO. En supposant que l'oxygène diffuse sous forme moléculaire, et en comparant la taille des molécules de CO et O2 on peut admettre que leurs coefficients de diffusion sont comparables. Si l'oxygène diffuse dans les lacunes, la diffusion du CO doit être beaucoup moins rapide mais cette situation est impossible.
- Si la réaction à l'interface est limitante, les gradients des pressions de CO et O2 sont faibles sauf à l'interface. Il faudrait que la diffusion soit beaucoup plus rapide que la réaction à l'interface. Or il est impossible que la diffusion à travers la couche d'oxyde soit la plus rapide.
- Si l'étape limitante est la diffusion de CO vers l'extérieur, la pression d'oxygène sera celle de l'atmosphère ambiante, soit 105Pa. La pression de CO sera suffisamment forte pour engendrer des fissures. La diffusion de CO à travers la couche d'oxyde ne peut pas être l'étape limitante.
La situation réelle doit être un régime mixte de diffusion d'oxygène et de CO.
La perméabilité de la protection au CO doit être très supérieure ou comparable à celle de l'oxygène pour empêcher la formation de bulle dans le dépôt.
3. CARACTERISTIQUES DES MATERIAUX
Les performances des composites HfB2-SiC ont été évaluées sur deux types d’échantillons, des monolithes et des revêtements élaborés sur différents substrats. Les conditions d’élaboration sont identiques, seuls les post traitements varient d’un échantillon à l’autre. Le comportement des matériaux bruts de siliciuration, post traités sous argon et sous azote seront présentés. Les caractéristiques physiques et chimiques des matériaux sont résumées dans le tableau 19.
Compte tenu de l’impossibilité de réaliser des mesures quantitatives sur les revêtements, nous supposerons que la microstructure obtenue est identique à celle des monolithes. Les principales différences rencontrées lors de l’élaboration sont dues à la formation d’une couche interfaciale de SiC entre le revêtement et le substrat et le développement de contraintes thermiques liées à la différence de coefficient de dilatation thermique, en particulier dans le cas des composites carbone/carbone et carbone/carbure de silicium. Des observations au MEB montrent que les grains de HfB2 ont une taille similaire pour les monolithes et les revêtements.
Tableau 19 : Caractéristiques des échantillons
4. PROTOCOLE EXPERIMENTAL
Les tests d’oxydation ont été réalisés à une pression totale de 105Pa, dans une thermobalance (setaram B60) reliée à un four 2073K et un tube laboratoire en alumine. Afin de limiter les interactions entre les échantillons et la nacelle, et de faciliter l’écoulement gazeux, les échantillons sont supportés par des tiges de HfO2 et placés dans une nacelle d‘alumine cylindrique, ouverte des deux côtés. La nacelle est suspendue dans la zone isotherme du four. La température est mesurée par un thermocouple (Pt 30%Rh / Pt 6%Rh).
4.1 Oxydation en montée linéaire de température
Deux types d’essais ont été entrepris, une montée linéaire à 8.33.10-2°/s jusqu’à 1873K sous un flux constant de gaz de 0,83.10-6 m3/s pour déterminer la température de début d’oxydation des composites ou une montée linéaire à 8.33.10-2°/s jusqu’à 1773K suivi d’un ralentissement de la vitesse à 1,66.10-2°/s jusqu’à 1973K pour déterminer avec précision la température de transition entre les domaines d’oxydation active et passive. Le flux de gaz est alors de 5.10-6 m3/s, afin de ne pas limiter la réaction par l’apport de gaz, sous de faibles pressions partielles d’oxygène. Les mélanges O2/He utilisés sont soit des gaz commerciaux (pression partielle d’oxygène de 2.104 Pa, 5.103 Pa ou 2.103 Pa), soit obtenus en mélangeant ces gaz à de l’hélium de qualité GC.
Après oxydation, les échantillons sont analysés par diffraction des rayons X, microsonde X et observation de sections polies au MEB. Les difficultés rencontrées pour déterminer la composition minérale ne permettent pas le calcul précis du degré d’avancement de la réaction. La variation de masse sera donc simplement pondérée par la surface géométrique de l’échantillon afin d’obtenir des résultats comparables.
4.2 Oxydation en régime isotherme
La montée en température est réalisée sous atmosphère inerte statique d’argon C. Le gaz oxydant est introduit rapidement, une fois la température de palier stabilisée. Après remplissage, le débit est ramené à 0,83.10-6 ou 5.10-6 m3/s et ajusté jusqu’à sa stabilisation. La durée du palier est fixée à 36000 s.
5. RESULTATS
5.1 Influence de la température
5.1.1 Montée linéaire de température
L’étude en montée linéaire de température a pour objectifs d’analyser l’influence du traitement thermique post siliciuration sur le comportement des matériaux et de mettre en évidence les différentes étapes de l’oxydation des composites HfB2-SiC, afin de les comparer avec des études antérieures. Ces mesures ont été réalisées uniquement sur les monolithes, compte tenu du nombre d’essais à réaliser. Les analyses thermogravimétriques ont été effectuées sous des pressions partielles d’oxygène de 5.103 et 2.104 Pa, sur des échantillons bruts de siliciuration, post traités sous argon, à 1973K pendant 7200s ou sous azote à 1823K pendant 14400s.
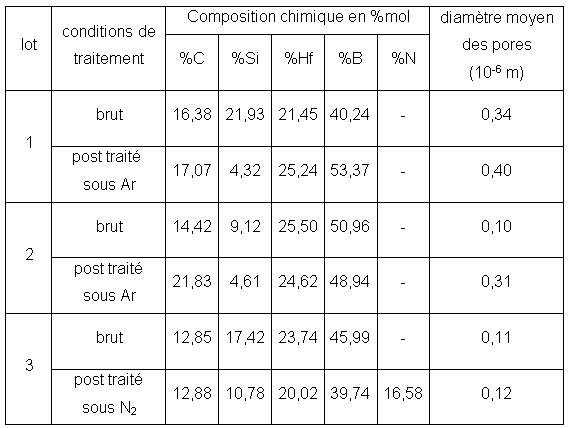
Les variations de masse montrent un gain très rapide qui débute entre 873-973K et se bloque rapidement entre 1273K et 1373K suivant la pression partielle d’oxygène et le traitement de l’échantillon (figure 57, figure 58, figure 59 et figure 60). Les courbes représentant la vitesse d’oxydation montrent l’existence de deux domaines. Le premier se situe entre 873K et 1023K et correspond à l’oxydation de la phase interfaciale contenant Hf-B-Si-C comme le montrent les analyses des produits de réaction (cf 5.3). Le second, entre 1073K et 1273K peut être attribué à l’oxydation des nanoparticules de SiC et de la phase Hf-B-Si-C. Les échantillons bruts de siliciuration présentent une vitesse d’oxydation plus élevée dans le premier domaine.
Le post traitement sous argon augmente la contribution de l’oxydation de SiC, qui peut devenir plus importante que celle associée à la phase interfaciale. Le gain de masse global est systématiquement plus important après post traitement. Ce comportement peut être expliqué par l’augmentation du volume poreux, qui nécessite une quantité plus importante de phase vitreuse pour former une couche protectrice.
Le post traitement sous azote entraîne un décalage du début de la réaction vers les plus hautes températures mais la vitesse de la réaction reste plus élevée dans le premier domaine (figure 59 et figure 60). Le gain de masse global est également supérieur mais dans une moindre mesure que dans le cas du post traitement sous argon, en particulier à basse pression partielle d’oxygène. La présence de grains de HfB2 et de la solution solide HfN1-xB2x pourrait expliquer ce décalage de la température de début d’oxydation. La formation d’une quantité plus importante de phase vitreuse à basse température favorise une cicatrisation rapide.
Figure 57 : Analyse thermogravimétrique et courbes dérivées de l’oxydation en montée linéaire sous un flux de 0,83.10-6 m3/s de mélange O2/He avec PO2=2.104Pa, de monolithes post traités ou non sous argon à 1973K pendant 7200s.
Figure 58 : Analyse thermogravimétrique et courbes dérivées de l’oxydation en montée linéaire sous un flux de 0,83.10-6 m3/s de mélange O2/He avec PO2=5.103Pa, de monolithes post traités ou non sous argon à 1973K pendant 7200s.
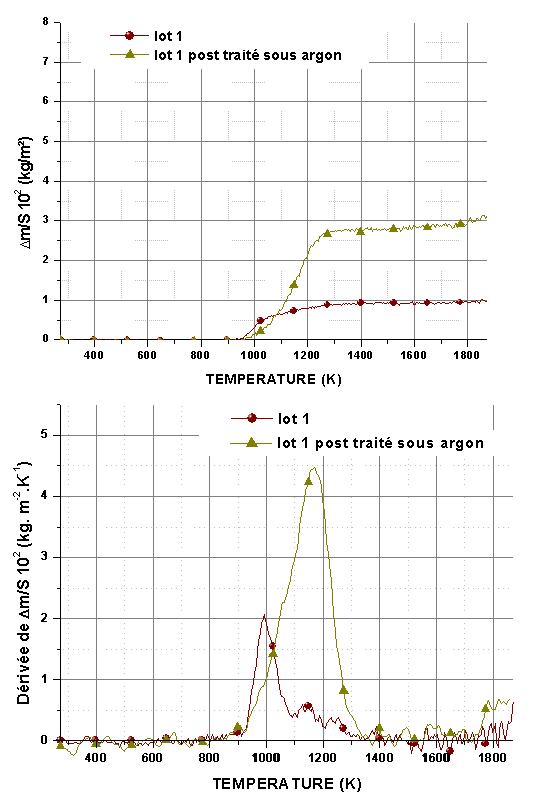
Figure 59 : Analyse thermogravimétrique et courbes dérivées de l’oxydation en montée linéaire sous un flux de 0,83.10-6 m3/s de mélange O2/He avec PO2=2.104Pa, de monolithes post traités ou non sous azote à 1823K pendant 14400s.
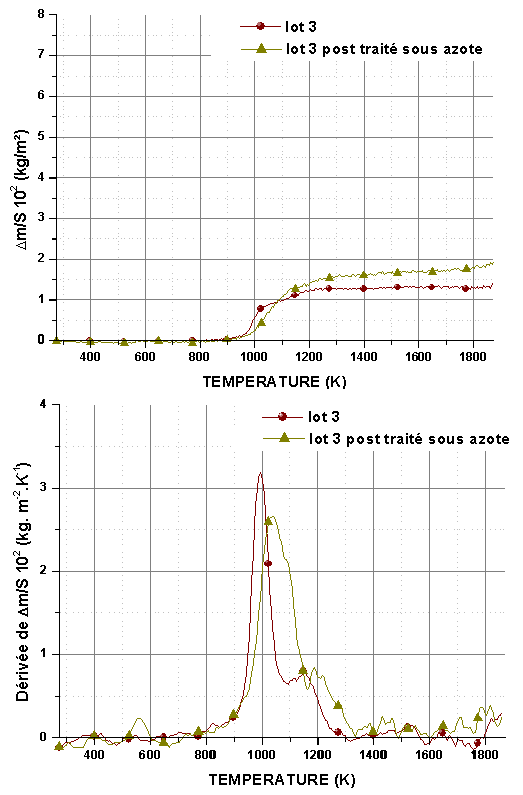
Figure 60 : Analyse thermogravimétrique et courbes dérivées de l’oxydation en montée linéaire sous un flux de 0,83.10-6 m3/s de mélange O2/He avec PO2=5.103Pa, de monolithes post traités ou non sous azote à 1823K pendant 14400s.
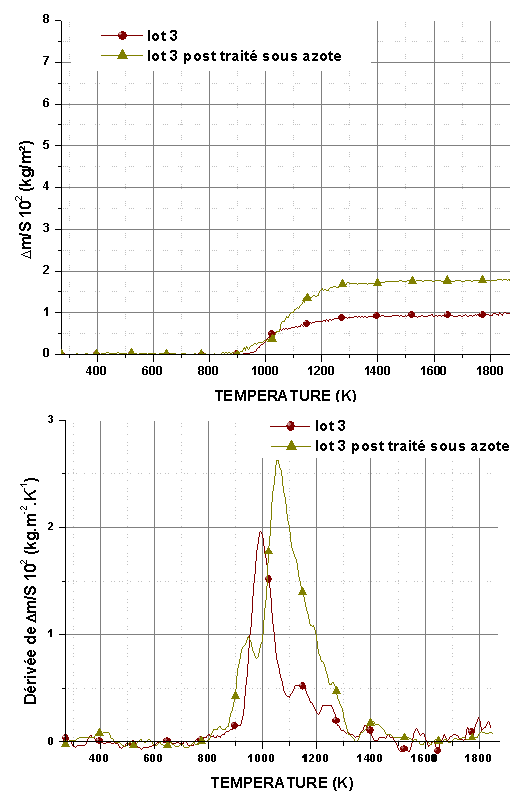
5.1.2 Oxydation en régime isotherme
L’objectif est d’étudier la cinétique d’oxydation en fonction du temps dans l’intervalle 1073-1973K, sous des pressions partielles d’oxygène comprises entre 102 et 2.104 Pa afin de prévoir la durée d’utilisation du revêtement.
Les mesures ont été effectuées sur les monolithes des mêmes lots que ceux utilisés pour la montée linéaire en température. Les cinétiques d’oxydation des revêtements ont été déterminées jusqu’à 1973K, sous des pressions partielles d’oxygène de 5.102 et 2.104 Pa afin de se rapprocher des limites d’utilisation.
Les isothermes ont été réalisées dans une large gamme de température afin de mettre en évidence les différents domaines et d’étudier la stabilité des produits de réaction ou l’évolution de leur composition en fonction du temps et de la température.
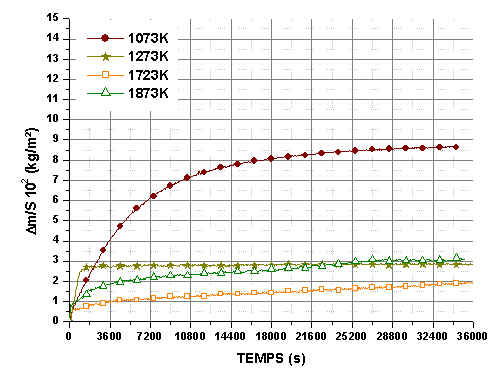
Pour les échantillons siliciurés, la variation de masse est très rapide pendant les premières minutes et atteint une asymptote quelle que soit la température testée. Après un palier de 36000s la valeur de Δm/S est la plus élevée pour 1073K puis elle diminue pour augmenter à nouveau. Ce comportement confirme l’existence de plusieurs mécanismes d’oxydation mis en évidence lors de la montée linéaire. Aucune perte de masse n’a été observée quelle que soit la température, sous une pression partielle d’oxygène de 2.104Pa. Après quelques heures de palier, la variation de masse est très faible, ce qui fait penser à un régime diffusionnel très lent ou à une compensation entre le gain de masse, dû à l’oxydation, et la perte de masse, liée à l’évaporation de la phase oxydée et un départ d’espèces volatiles.
Le gain de masse total est systématiquement supérieur pour les échantillons post traités sous argon, avec une accélération de la cinétique de la réaction au bout de 14400 s, à 1723K et 1873K (figure 61 et 62). L’élargissement de la porosité peut expliquer que le temps nécessaire a son comblement augmente.
Figure 61 : Isothermes d’oxydation de monolithes du lot 1, post traités à 1973K/7200s sous argon et non post traités, sous le mélange O2/He avec PO2=2.104Pa et un débit de 0,83.10-6 m3/s
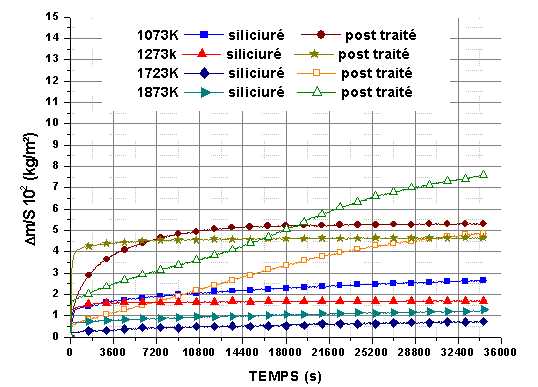
Figure 62 : Isothermes d’oxydation de monolithes du lot 2, post traités à 1973K/7200s sous argon et non post traités, sous le mélange O2/He avec PO2=2.104Pa et un débit de 0,83.10-6 m3/s
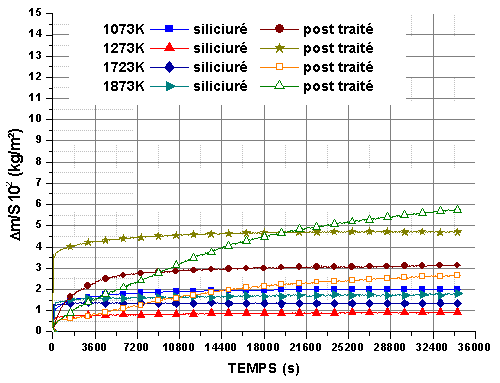
Les mêmes isothermes ont été réalisées sur des monolithes post traités sous azote pour des pressions partielles d’oxygène de 5.103 et 2.104 Pa (figure 63 et figure 64). Comme dans le cas précédent, aucune perte de masse n’est observée et la réaction atteint rapidement un palier.
A basse température, la diminution de la pression partielle d’oxygène ne semble pas modifier la cinétique de la réaction. A nouveau, le gain de masse est important à 1073K, mais inférieur à celui enregistré sous 2.104Pa, ce qui indique que la réaction dépend de la pression partielle d’oxygène et de la formation de la phase cicatrisante. Ce n’est pas le cas à toutes les autres températures.
Le post traitement sous azote améliore la stabilité à haute température et sous faible pression partielle d’oxygène. On n’observe pas d’accélération de la réaction après 14400 ou 18000 s mais une décroissance monotone de la vitesse.
Figure 63 : Isothermes d’oxydation de monolithes du lot 3, post traités à 1823K/14400s sous azote, sous le mélange O2/He avec PO2=2.104Pa et un débit de 0,83.10-6 m3/s
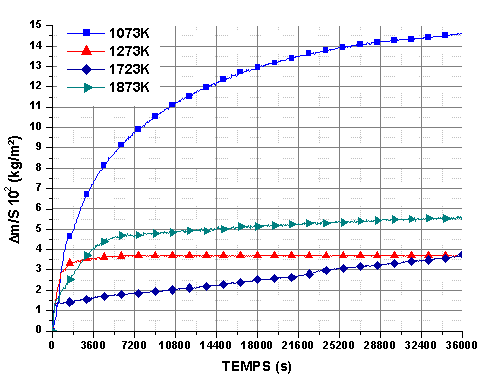
Figure 64 : Isothermes d’oxydation de monolithes du lot 3, post traités à 1823K/14400s sous azote, sous le mélange O2/He avec PO2=5.103Pa et un débit 0,83.10-6 m3/s
Les variations de masse des échantillons conduisent dans la plupart des cas à un palier. Cependant, afin de savoir si la vitesse d’oxydation est très faible ou s’il y a compensation entre gain et perte de masse liées aux réactions, nous avons étudié la variation de l’épaisseur oxydée avec le temps sur des monolithes, post traités sous azote. L’épaisseur des monolithes a été mesurée par observation au microscope optique d’une section polie, avant et après oxydation et après polissage de la section pour faire apparaître l’épaisseur de la couche oxydée
Des isothermes à 1873K sous une pression partielle de 2.104Pa, pendant 10800 s, 18000 s et 36000 s ont été réalisées (figure 65). Elles montrent dans un premier temps une bonne reproductibilité de la variation de masse totale. L’épaisseur de la couche oxydée semble varier très peu avec le temps, comme le montrent les valeurs moyennes sur 20 mesures (tableau 20) et quelle que soit l’épaisseur initiale du monolithe, ce qui correspond aux observations des sections polies sur un autre lot (figure 66).
Ces valeurs confirment que la cinétique de la réaction est très lente à 1873K sous une pression partielle d’oxygène de 2.104Pa et que l’épaisseur oxydée est assez homogène sur toute la surface de l’échantillon.
Figure 65 : Isothermes à 1873K sous 2.104Pa de mélange O2/He de monolithes du lot 3, post traités sous azote à 1823K/14400s débit 0,83.10-6 m3/s
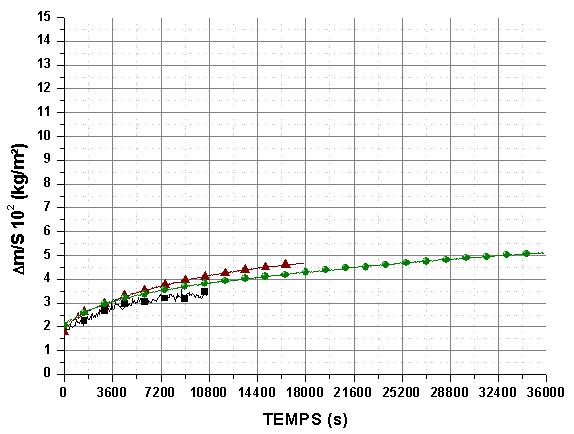
Figure 66 : Observation au MEB d’une section polie de monolithe, post traité sous azote, après oxydation à 1873K pendant 36000s sous un mélange O2/He avec PO2=2.104Pa

5.2 Influence de la pression partielle d’oxygène
Le revêtement pourrait être utilisé sous faible pression partielle d’oxygène, par exemple dans des conditions de rentrée atmosphérique. Lors de la phase de descente, les températures les plus importantes sont rencontrées dans les hautes couches de l’atmosphère, où la pression partielle d’oxygène est faible (103 Pa) puis cette dernière augmente progressivement.
La détermination du domaine d’oxydation active ou passive a une grande importance pour prévoir la gamme d’utilisation du matériau. La transition a été évaluée sur les monolithes et les revêtements et comparée à celle de fibres de carbure de silicium (Nicalon NLM 202) et de poudre de SiC (Alfa 99,8% SiC-β) (figure 67). Les fibres sont constituées de nanocristaux de SiC dont la taille est assez proche de celle du SiC présent dans le composite HfB2-SiC.
Figure 67 : Température de transition entre les domaines d’oxydation active et passive pour les fibres Nicalon NLM-202, de la poudre de SiC (99,8% SiC-β) et les composites HfB2-SiC. Comparaison avec le carbure de silicium(1)
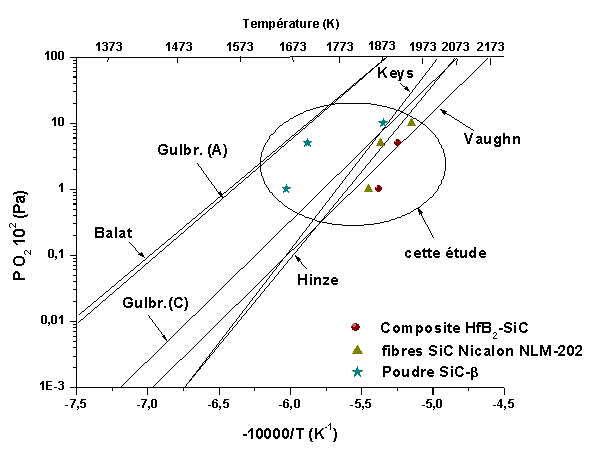
Les montées linéaires de température sont réalisées à des vitesses de 8.33.10-2°/s jusqu’à 1773K puis 1,66.10-2°/s jusqu’à 1973K, afin d’améliorer la précision sur la détermination de la température de transition. Le débit de gaz est de 5.10-6 m3/s pour ne pas limiter la cinétique par la diffusion dans la couche limite. Les pressions partielles d’oxygène sont comprises entre 102, 5.102 et 103 Pa.
Pour les composites HfB2-SiC, le domaine d’oxydation passive paraît légèrement plus étendu par rapport aux fibres Nicalon. Sous une pression partielle d’oxygène de 103Pa, la transition n’est pas atteinte à 1973K, limite en température du four de la thermobalance.
Par ailleurs la vitesse de montée en température a une influence sur la transition. Ainsi, une vitesse constante de 8.33.10-2°/s la décale vers les plus hautes températures, car sous une pression partielle d’oxygène de 102Pa, la température de transition passe de 1860K à 1914K.
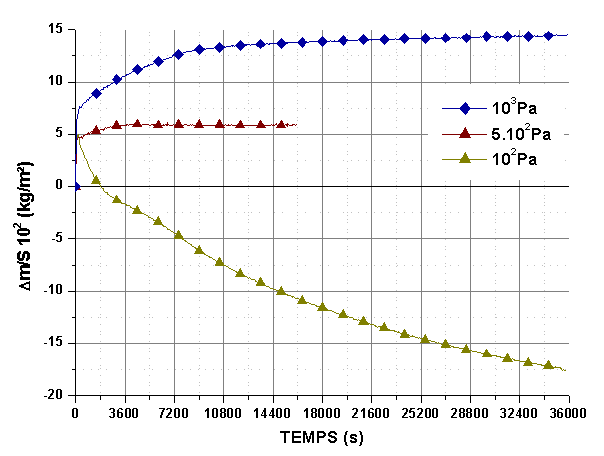
Le comportement à basse pression partielle d’oxygène a été précisé sur des monolithes et sur des revêtements à 1873K et 1973K. Les isothermes de la figure 68 confirment que la transition entre les domaines d’oxydation active et passive a lieu entre des pressions partielles d’oxygène de 102 et 5.102 Pa.
Figure 68 : Isothermes à 1873K de monolithes du lot 3 post traités sous azote, sous des pressions partielles d’oxygène de 102, 5.102 et 103Pa pendant 36000s avec un débit 5.10-6 m3/s
Le substrat joue un rôle dans le développement des contraintes thermiques et donc de la fissuration du revêtement. L’efficacité de la protection de différents substrats a été étudiée à très haute température sous des pressions partielles d’oxygène de 5.102 et 2.104Pa, et un débit de 0,83.10-6 m3/s.
Les variations de masse montrent la formation d’une couche oxydée compacte et protectrice sous 2.104Pa (figure 69, figure 70, figure 71). La perte de masse enregistrée à 1973K après 14400 s est due à la formation d’une piqûre dans le revêtement (figure 69). Le domaine d’oxydation active semble être atteint sous 5.102Pa car l’échantillon ne semble pas recouvert d’une phase vitreuse (figure 72). La protection est néanmoins efficace car ni écaillage, ni attaque locale du substrat n’ont été observés. L’analyse thermogravimètrique montre qu’après un faible gain de masse, la perte tend vers une asymptote, ce qui confirme le caractère protecteur du revêtement, malgré son endommagement. La partie externe du revêtement devient poreuse mais toute l’épaisseur n’est pas attaquée. Si l’on calcule le degré d’avancement de l’attaque du revêtement en supposant que la composition est 80%volume HfB2 et 20%volume SiC, il est de 20 à 25% après 36000s à 1873K, ce qui confirme le caractère protecteur de la partie non oxydée du revêtement.
Figure 69 : Revêtements élaborés sur graphite. Isothermes à 1873K et 1973K sous 5.102 ou 2.104Pa d’oxygène avec un débit de 0,83.10-6 m3/s
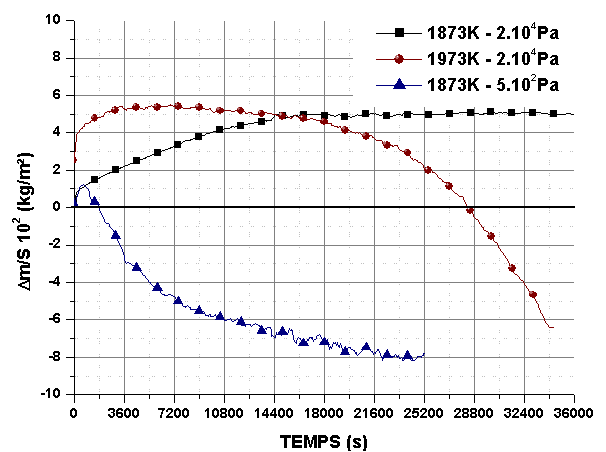
Figure 70 : Observation MEB d’une section polie (a et b) d’un revêtement sur graphite, oxydé à 1873K/36000s sous un mélange O2/He avec une pression partielle d’oxygène de 2.104Pa et un débit de 0,83.10-6 m3/s et photo optique (c)

Figure 71 : Observation MEB d’une section polie (a et b) d’un revêtement sur graphite, oxydé à 1973K/36000s sous un mélange O2/He avec une pression partielle d’oxygène de 2.104Pa et un débit de 0,83.10-6 m3/s et photo optique (c)

Figure 72 : Observation MEB d’une section polie d’un revêtement sur graphite (a et b), oxydé à 1873K/36000s sous un mélange O2/He avec une pression partielle d’oxygène de 5.102Pa et un débit de 0,83.10-6 m3/s et photo optique (c)

Les composites carbone/carbure de silicium ont un coefficient de dilatation thermique inférieur à celui du graphite et anisotrope en fonction de l’orientation des fibres de carbone, ce qui peut favoriser la formation d’un réseau de fissures dans le revêtement.
L’isotherme à 1873K sous une pression partielle d’oxygène de 2.104Pa montre la protection du revêtement pendant 7200 s (figure 73). A la sortie du four, le revêtement est écaillé au niveau des arêtes, ce qui explique la perte de masse qui se poursuit jusqu’à la fin du palier. Les observations des sections polies, dans les zones n’ayant pas subi d’écaillage, montrent la formation d’une couche d’oxyde protectrice d’environ 20.10-6 m (figure 74 et figure 75). Le comportement du revêtement élaboré sur les composites est donc similaire à ceux élaborés sur graphite.
La géométrie de l’éprouvette est un paramètre important. Les arêtes des substrats composites sont des zones de concentration de contraintes, qui conduisent à un déchirement du revêtement. Des pièces avec des rayons de courbure beaucoup plus grands auraient été plus appropriées.
Figure 73 : Isothermes à 1873K et 1973K sous 2.104Pa avec un débit de 0,83.10-6 m3/s, de revêtements élaborés sur composite C/SiC
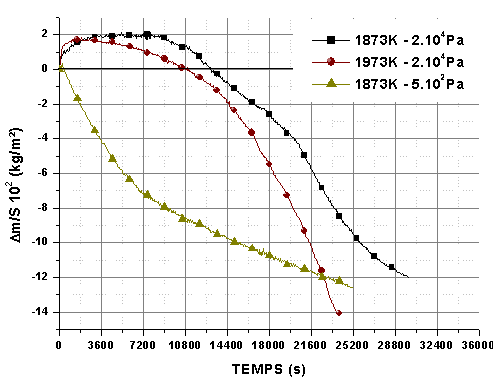
figure 74 : Observation MEB (a et b) d’une section polie d’un revêtement sur composite C/SiC, oxydé à 1873K/36000s sous un mélange O2/He avec une pression partielle d’oxygène de 2.104Pa et un débit de 0,83.10-6 m3/s et photo optique (c)

Figure 75 : Observation MEB d’une section polie d’un revêtement sur composite C/SiC, oxydé à 1973K/36000s sous un mélange O2/He avec une pression partielle d’oxygène de 2.104Pa et un débit de 0,83.10-6 m3/s

5.3 Produits de la réaction
Les diagrammes de diffraction des rayons X (figure 76) réalisés sur des monolithes oxydés entre 1073K et 1873K sous une pression de 2.104Pa d’oxygène montrent la formation de HfO2 dès 1073K et la présence d’une phase vitreuse. Aucune autre phase cristallisée, issue de l’oxydation, n’est détectée, quelle que soit la température.
Afin de déterminer la composition et la répartition des produits de la réaction des cartographies élémentaires, des profils et des analyses locales de composition ont été entrepris par microsonde X.
Les échantillons observés sont des sections polies soit de monolithes, soit de revêtements, oxydés en régime isotherme.
Après 36000s à 1073K, sous une pression d’oxygène de 2.104Pa (figure 77), la profondeur de pénétration de l’oxygène est importante, malgré la formation d’une couche de silice protectrice. La cinétique de croissance de cette couche semble être lente à basse température, permettant la diffusion d’une petite quantité d’oxygène sur une grande distance avant le comblement de la porosité ouverte par l’oxyde.
Après 86400s à 1723K, sous une pression d’oxygène de 2.104Pa (figure 78), une couche cicatrisante d’environ 10.10-6 m d’épaisseur, riche en silice et de faible teneur en bore, est formée à la surface de l’échantillon. La pénétration de l’oxygène est limitée à la couche d’oxyde.
Le comportement en régime isotherme à 1873K a été évalué à 5.102 et 2.104Pa (figure 79 et figure 80). Comme montré dans le paragraphe 5.2, ces pressions partielles d’oxygène correspondent respectivement à une oxydation active et passive du matériau. Sous 5.102Pa d’oxygène, il ne se forme pas de couche protectrice. Les cartographies par microsonde montrent que le carbure de silicium est oxydé préférentiellement (figure 80).
Sous 2.104Pa, une couche d’oxyde d’environ 20.10-6 m d’épaisseur se forme à la surface de l’échantillon. Des analyses locales et des profils de concentration ont mis en évidence la présence d’un gradient de bore entre les interfaces interne et externe de la couche d’oxyde, indiquant un départ de composés contenant du bore par diffusion. De plus, la phase vitreuse contient du hafnium (tableau 21).
| Elément | Teneur moyenne (%atomique) |
| O | 70,4 |
| Si | 28,5 |
| B | 0,57 |
| Hf | 0,52 |
Après 36000s à 1973K, sous 2.104Pa d’oxygène (figure 81), la phase vitreuse externe est riche en silice et contient environ 0,5%atomique d’hafnium (tableau 22). La microstructure est très proche de celle obtenue à 1873K, ce qui confirme la stabilité de la phase vitreuse à très haute température. On notera la teneur importante en bore à proximité de l’interface entre le matériau non oxydé et la couche d’oxyde (mesure 1 tableau 22)
Figure 76 : Diagramme de diffraction des rayons X de la surface de monolithes oxydés pendant 36000s sous une pression partielle d’oxygène de 2.104Pa
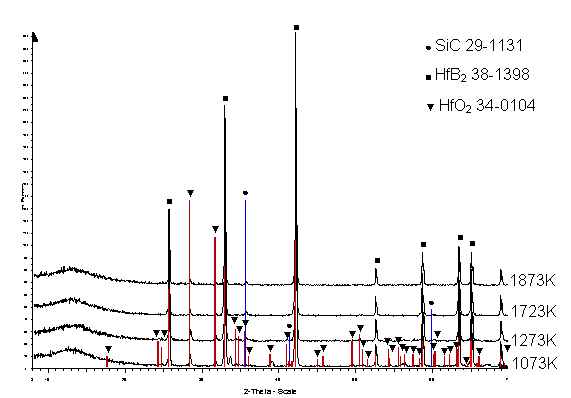
Figure 77 : Analyse à la microsonde X d’une section polie de monolithe, post traité sous azote, après oxydation à 1073K/36000s sous un mélange O2/He avec PO2=2.104Pa

Figure 78 : Analyse à la microsonde X d’une section polie de monolithe, post traité sous azote, après oxydation à 1723K/86400s sous un mélange O2/He avec PO2=2.104Pa

Figure 79 : Analyse à la microsonde X d’une section polie de monolithe, post traité sous azote, après oxydation à 1873K/36000s sous un mélange O2/He avec PO2=2.104Pa

Figure 80 : Analyse à la microsonde X d’une section polie de revêtement, post traité sous azote, après oxydation à 1873K/36000s sous un mélange O2/He avec PO2=5.102 Pa

Figure 81 : Analyse à la microsonde X d’une section polie de revêtement, post traité sous azote après oxydation à 1973K/36000s sous un mélange O2/He avec PO2=2.104Pa


5.4 Discussion et conclusions
Les comportements à l’oxydation des revêtements et des monolithes sont comparables dans les conditions testées. Les isothermes présentent un gain de masse pendant les premières minutes et un ralentissement rapide de la cinétique qui reste très lente pendant toute la suite du palier. Deux domaines de température peuvent être isolés d’un point de vue cinétique, en fonction des produits de la réaction.
A 1073K et 1273K les isothermes ont montré la formation d’un verre borosilicaté riche en B2O3, issu de l’oxydation de la phase intermédiaire située à la périphérie des grains de HfB2. La vitesse de formation et la viscosité de la phase vitreuse, ainsi que le volume de la porosité résiduelle contrôlent la profondeur de pénétration de l’oxygène avant d’atteindre un palier par formation d’une couche protectrice dense. A ces températures, la volatilisation d’espèces contenant du bore, à partir du borosilicate, est faible. L’augmentation de la viscosité liée à la présence de silice ne semble pas pénaliser le caractère protecteur à 1073K puisque la phase vitreuse recouvre l’ensemble de l’échantillon. Le traitement thermique post siliciuration ne modifie pas profondément la tenue du revêtement. On note un faible décalage du début de la réaction vers les hautes températures avec un gain de masse plus important lié à la porosité plus élevée. Nous avons essayé de décrire la cinétique d’oxydation à partir du modèle asymptotique développé par Evans(11) pour Si3N4 :
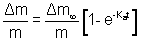
avec 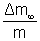 la prise de masse maximale
la prise de masse maximale
Ka la constante de temps
t le temps
Les principes de base qui ont conduit à ce modèle sont les suivants :
- La vitesse de réaction est déterminée par le nombre de pores restant ouverts
- Le blocage apparent de l’oxydation, après fermeture complète de la porosité, traduit le caractère protecteur de l’oxyde formé sur la surface géométrique de l’échantillon. La surface réactive évolue avec la température et le temps d’oxydation.
Dans les premiers instants, le film oxyde recouvre les surfaces internes liées à la porosité ouverte et la surface externe de l’échantillon. Ensuite, la couche formée entraîne la fermeture progressive de la porosité. L’étude n’est possible qu’à 1073K car le blocage est assez lent mais ce modèle ne décrit pas parfaitement le comportement observé sur les matériaux élaborés par infiltration réactive.
Le comportement de nos matériaux est très différent de celui observé sur les échantillons élaborés par frittage sous charge. Ces derniers subissent une oxydation préférentielle du borure, conduisant à la formation de B2O3 et de HfO2. La cinétique, régie par un régime diffusionnel, est similaire à celle du borure seul, avec une évaporation rapide de B2O3 avant que le carbure de silicium ne commence à s’oxyder. Hinze et al(12) ont mis en évidence la transition entre l’oxydation préférentielle du borure et le début de l’oxydation de SiC à environ 1623K.
Dans l’intervalle 1723-1973K, les cinétiques sont très lentes et le domaine affecté par l’oxydation est très inférieur à celui observé à plus basse température. Les analyses par microsonde mettent en évidence la présence d’une très faible quantité de bore et d’hafnium dans la phase vitreuse essentiellement composée de silice. Le départ des composés du bore s’effectue à travers la couche oxydée car aucune porosité ou bulle n’est observée.
Compte tenu de l’allure des isothermes et de la morphologie des produits d’oxydation nous avons calculé les transformées (Δm/S)²=f(t).
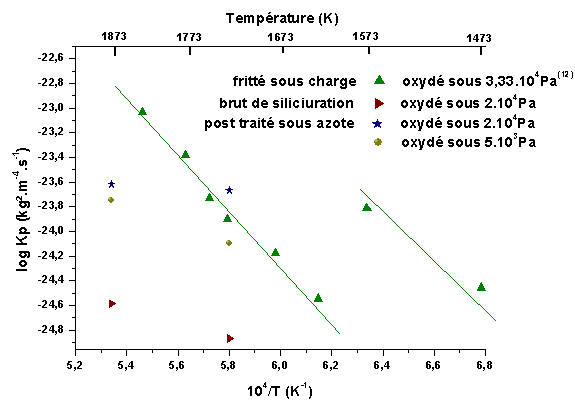
Les graphes de la figure 82 montrent qu’un régime de diffusion s’installe progressivement après quelques heures de palier. Les constantes de diffusion pour 1723K et 1873K, calculées pour les différents matériaux sont reportées sur la figure 83 et comparées aux valeurs obtenues par Hinze(12) sur des matériaux frittés. Le dégagement d’azote pour les échantillons post traités pourrait expliquer les constantes plus élevées par rapport aux échantillons bruts de siliciuration. On constate que les constantes de diffusion déterminées pour une température de 1873K sont inférieures pour les matériaux élaborés par siliciuration. De plus, on remarque, comme pour l’oxydation des composés à base de silicium, une influence de la pression d’oxygène sur la constante de diffusion, ce qui milite en faveur d’une migration d’oxygène limitante à travers le verre silicaté. Un facteur 2 apparaît entre les constantes mesurées sous des pressions d’oxygène de 2.104Pa et 5.103Pa
La meilleure résistance à l’oxydation des matériaux élaborés par infiltration réactive peut être attribuée à leur microstructure. La présence d’une phase Hf/B/Si/C en périphérie des grains de HfB2 et la plus grande réactivité des nanocristaux de SiC induisent la formation d’un borosilicate et une dissolution progressive de HfO2. Davis et al(10)ont montré que des oxydes de type Na2O, Al2O3, B2O3 améliorent la solubilité de HfO2 dans les verres borosilicatés, ce qui doit se traduire par une plus grande stabilité et une viscosité plus élevée. Ainsi, s’expliquerait la très bonne tenue à l’oxydation des matériaux à 1973K sous 2.104Pa et la diminution importante de la constante de diffusion kD au-dessus de 1723K, lorsque la teneur en HfO2 dans le silicate augmente. La diffusion de l’oxygène dans un verre plus visqueux serait plus lente.
Au dessus de 1873K et sous une faible pression partielle d’oxygène on constate un départ de silicium et d’oxygène comparable au phénomène d’oxydation active de SiC. Mais contrairement à l’oxydation du carbure, la vitesse de perte de masse se ralentit progressivement et tend vers une asymptote. Par analyses à la microsonde on met en évidence une phase vitreuse dans les pores entourant les grains de HfB2 et HfO2. Ainsi un revêtement oxydé à 1873K pendant 36000s sous une pression partielle de 5.102Pa conserve un caractère protecteur. L’épaisseur de la couche oxydée est de 50.10-6 m soit un degré d’avancement de la réaction de l’ordre de 20 à 25%.
Figure 82 : Transformées des cinétiques d’oxydation de monolithes HfB2-SiC, bruts de siliciuration, oxydés sous 5.103Pa d’oxygène
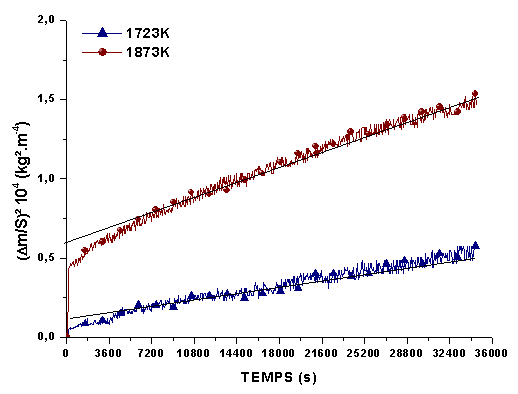
Figure 83 : Constantes de vitesse paraboliques pour l’oxydation de HfB2-SiC(0)
REFERENCES BIBLIOGRAPHIQUES
- BALAT, M. J. H. Determination of the active to passive transition in the oxidation of silicon carbide in standard and microwave-excited air. J. Eur. Ceram. Soc., 16, 1996, 55-62.
- BERKOWITZ-MATTUCK, J. B. High temperature oxidation. III Zirconium and Hafnium diborides. J. Electrochem. Soc., 1966, 908-914.
- BULL, J. D., RASKY, D. J., et al. Stability characterization of diboride composites under high velocity atmospheric flight conditions. 24th international SAMPE Technical Conference. 1992
- CLOUGHERTY, E. et ANTHONY, F., Research and development of refractory oxidation-resistant diborides. Part II Application evaluations and design considerations. Technical report AFML-TR-68-190, 7, 1970
- CLOUGHERTY, E., HILL, R. et al. Research and development of refractory oxidation-resistant diborides. Party II Processing and characterisation. Technical report AFML-TR-68-100, 2, 1970
- CLOUGHERTY, E., KALISH D., et al. Research and development of refractory oxidation resistant diborides. Technical report AFML-TR-68-190, 1968
- CLOUGHERTY, E., KENNETH E., et al. Research and development of refractory oxidation-resistant diborides. Part II Thermal, physical, electrical and optical properties. Technical report AFML-TR-68-100, 5, 1969
- COSTELLO J. A. et TRESSLER R. E., Oxidation kinetics of silicon carbide crystals and ceramics : I, In dry oxygen., J. Am. Ceram. Soc., 69, 9, 1986, 674-681
- DAVIS, J. B., MARSHALL D. B., et al. Ceramic composites for thermal protection systems. composite Part A, 30, 1999, 483-488.
- DAVIS, L. L., LI G. G., et al. The effects of Na2O, Al2O3 and B2O3 on HfO2 solubility in borosilicate glass. Mat. Res. Soc. Symp. Proc., 556, 1999, 313-320.
- EVANS, U. R. The corrosion and oxidation of metals, Edward Arnold. 1960
- HINZE, J. W. et TRIPP W. C., The high temperature oxidation behavior of a HfB2+20v/o SiC composite. J. Electrochem. Soc., 122, 9, 1975, 1249-1254.
- KAUFMAN, L. Borides composites - a new generation of nose cap and leading edge materials for reusable lifting re-entry systems. AIAA Advanced Space Transportation Meeting, Cocoa Beach, Florida. 1970
- KAUFMAN, L., CLOUGHERTY E. V., et al. Oxidation characteristics of hafnium and zirconium diboride. Trans. of the metal. Soc. of AIME, 239, 1967, 458-466.
- LEVINE, S. R., OPILA E. J., et al. Evaluation of ultra-high temperature ceramics fora eropropulsion use. J. Eur. Ceram. Soc., 22, 14-15, 2002, 2757-2767.
- LUTHRA, K. Oxidation of carbon-carbon composites- a theoretical analysis. Carbon, 26, 2, 1988, 217-224.
- LUTHRA K. L., Some new perspectives on oxidation of silicon carbide and silicon nitride., J. Am. Ceram. Soc., 74, 5, 1991, 1095-1103
- NARUSHIMA T., GOTO T. et HIRAI T., High-temperature passive oxidation of chemically vapor deposited silicon carbide., J. Am. Ceram. Soc., 72, 8, 1989, 1386-90
- TRIPP, W. C. Thermogravimetric study of the oxidation of ZrB2 in temperature range of 800° to 1500°C. J. Electrochem. Soc., 118, 7, 1971, 1195-1199.
- TRIPP, W. C., DAVIS H. H., et al. Effect of an SiC addition on the oxidation of ZrB2. Ceram. Bull., 52, 1973, 612-616.
- VAUGHN W. L., Active-to-passive transition in the oxidation of silicon carbide and silicon nitride in air., J. Am. Ceram. Soc.,73, 6, 1990, 1540-1543